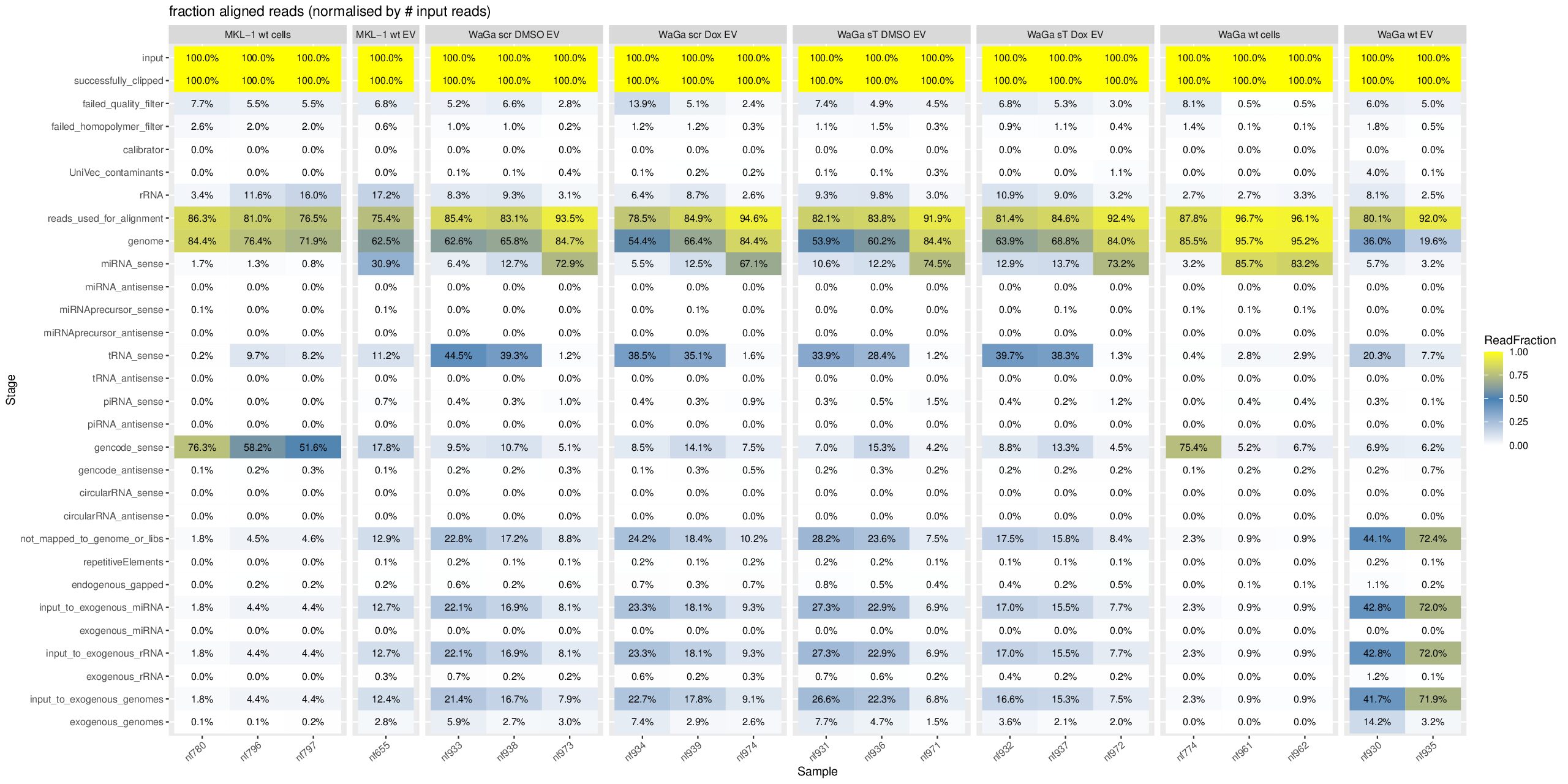
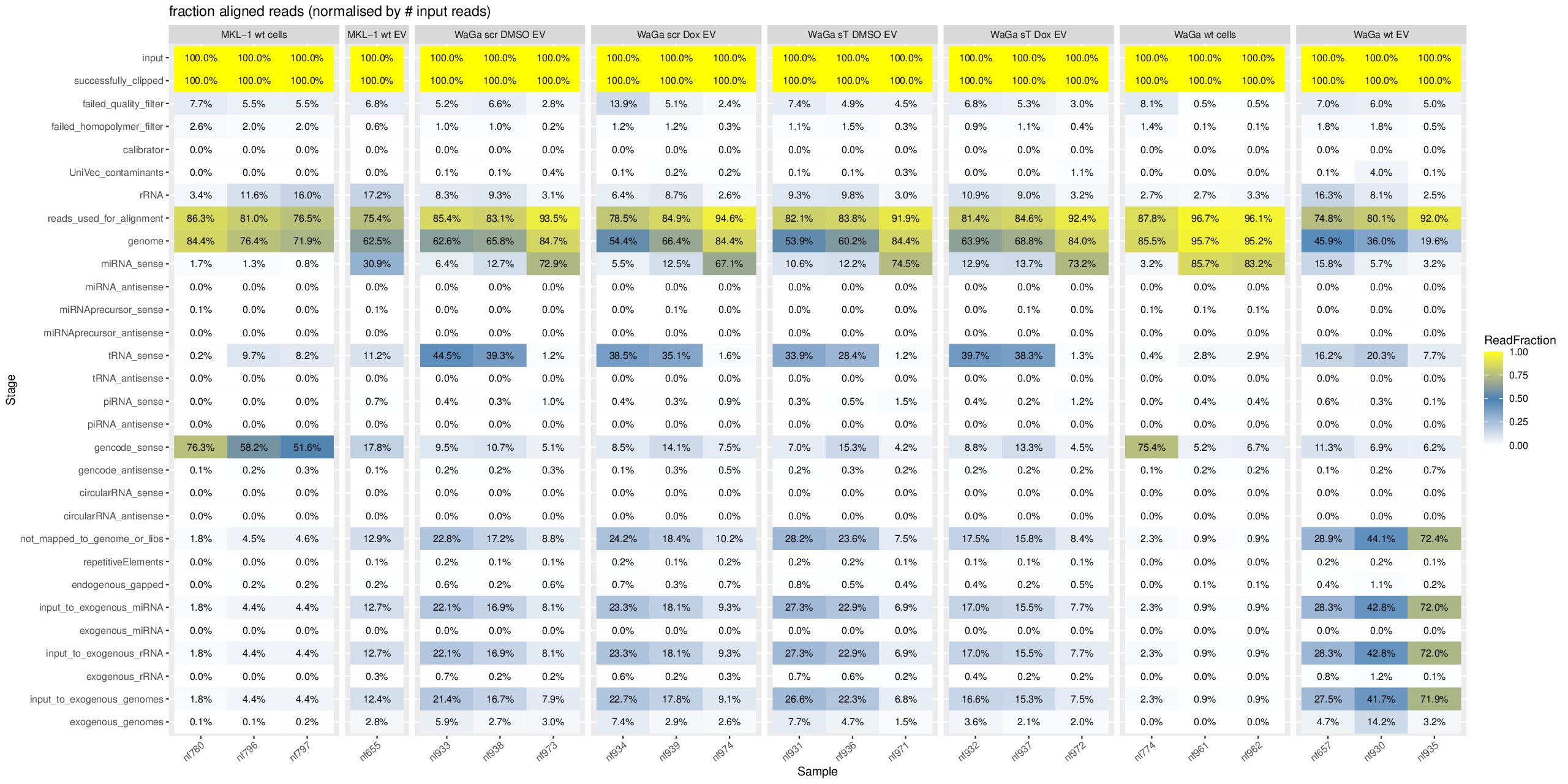
exceRpt流程热图分析(中文翻译)
行标签详细说明
根据您提供的mapping_heatmap3.pdf热图,以下是各类别的详细解释:
1. 外源性序列(Exogenous Sequences)
- exogenous_genomes: 比对到外源基因组(病毒/细菌)的reads
- exogenous_rRNA: 比对到外源rRNA的reads
- exogenous_miRNA: 比对到外源miRNA的reads
2. 内源性基因组特征
- genome: 比对到参考基因组的reads(这是主要类别)
- endogenous_gapped: 需要间隔比对的内源性reads(剪接reads)
- repetitiveElements: 比对到重复序列的reads
- not_mapped_to_genome_or_libs: 未比对到基因组或小RNA文库的reads
3. 编码/非编码RNA(GENCODE)
- gencode_sense: 比对到GENCODE注释基因的正链reads(mRNA、lncRNA等)
- gencode_antisense: 反义链reads
4. 小RNA类别
- miRNA_sense: 成熟miRNA正链(重要类别)
- miRNA_antisense: 成熟miRNA反义链
- miRNAprecursor_sense: miRNA前体正链
- miRNAprecursor_antisense: miRNA前体反义链
- tRNA_sense/antisense: 转运RNA
- piRNA_sense/antisense: PIWI互作RNA
- circularRNA_sense/antisense: 环状RNA
不同组别的主要内容变化趋势
📊 关键发现:
1. 基因组比对效率(genome行)
- MKL-1 wt: 82.9%, 61.4%, 97.4% – 变异很大
- MKL-1 wt EV: 97.9% – 非常高
- WaGa wt: 99.0%, 99.0%, 91.9% – 最高且稳定
- WaGa wt EV: 91.0%, 90.6%, 89.2% – 略低于wt
趋势: WaGa野生型样本显示最高的基因组比对效率(91-99%)
2. miRNA含量(miRNA_sense行)- 🔴 最重要发现
- MKL-1 wt: 41.0%, 21.1%, 3.7% – 剧烈变化
- MKL-1 wt EV: 1.9% – 极低
- WaGa scr DMSO EV: 1.6%, 1.0%, 7.1% – 低miRNA
- WaGa wt: 88.7%, 86.6%, 81.1% – 异常高的miRNA!
- WaGa wt EV: 79.3%, 77.9%, 70.9% – 仍然非常高
关键发现: WaGa野生型细胞的miRNA含量是MKL-1的20-40倍!这表明细胞系之间存在根本性的生物学差异。
3. 未比对reads(not_mapped_to_genome_or_libs)
- MKL-1 wt: 14.8%, 21.7%, 0.5% – 变异大
- WaGa wt: 2.9%, 3.0%, 1.3% – 最低
- WaGa wt EV: 1.4%, 1.2%, 1.7% – 非常低
趋势: WaGa样本总体比对效率更好(<3%未比对)
4. gencode(mRNA/lncRNA)
- MKL-1 wt: 0.0-1.0% – 极低
- WaGa wt: 0.4%, 0.5%, 1.6% – 较高
- WaGa wt EV: 1.4%, 1.1%, 0.9% – EV中mRNA升高
5. 外源性内容
- 所有组: exogenous_miRNA, exogenous_rRNA, exogenous_genomes均为0.0%
✅ 好消息: 所有样本均未检测到病毒或细菌污染
🎯 生物学意义总结
1. 细胞系特异性miRNA谱
- WaGa细胞: miRNA丰富(70-89%的reads)
- MKL-1细胞: miRNA贫乏(2-41%的reads)
- 意义: WaGa可能是更好的miRNA研究模型
2. EV vs 细胞RNA
- MKL-1 wt EV: miRNA降低(1.9%)vs 细胞(3.7-41%)
- WaGa wt EV: 维持高miRNA(71-79%)vs 细胞(81-89%)
- 解释: WaGa细胞优先将miRNA包装到EV中
3. 质量控制
- 最佳质量: WaGa wt和WaGa wt EV(低未比对,高基因组比对)
- 变异质量: MKL-1 wt(样本间变异大)
4. 建议
- 调查MKL-1变异: 为什么nf796只有21% miRNA而nf780有41%?
- WaGa作为miRNA模型: WaGa细胞更适合miRNA/EV-miRNA研究
- EV包装机制: WaGa细胞显示高效的miRNA包装到EV中 – 值得进一步研究
TODO: 进行统计分析或创建可视化图表来突出这些趋势!
Detailed Explanation of exceRpt Pipeline Row Labels
Based on your heatmap from the exceRpt pipeline, let me explain each category and specifically address your question about the miRNA percentages.
Pipeline Flow Overview
The exceRpt pipeline processes reads through sequential filtering and alignment stages. Each percentage is normalized to reads_used_for_alignment (the denominator after quality control).
Row Label Categories Explained
1. Initial Processing Stages
- input: Total raw reads entering the pipeline (100%)
- successfully_clipped: Reads with adapters successfully removed
- failed_quality_filter: Reads failing quality thresholds (Phred score, length)
- failed_homopolymer_filter: Reads with excessive homopolymer stretches
- calibrator: Reads mapping to spike-in calibrator controls
- UniVec_contaminants: Reads identified as vector/contaminant sequences
- rRNA: Reads mapping to ribosomal RNA (typically removed)
- reads_used_for_alignment: Clean reads proceeding to alignment (this is the new 100% for downstream percentages)
2. Endogenous Alignment (Human/Host Genome)
These reads map to the reference genome of the host organism:
- genome: Reads mapping to the reference genome
- miRNA_sense/antisense: Reads mapping to endogenous miRNAs in sense/antisense orientation
- miRNAprecursor_sense/antisense: Reads mapping to miRNA precursor hairpins
- tRNA_sense/antisense: Transfer RNA mappings
- piRNA_sense/antisense: PIWI-interacting RNA mappings
- gencode_sense/antisense: Reads mapping to GENCODE gene annotations (mRNA, lncRNA, etc.)
- circularRNA_sense/antisense: Circular RNA (circRNA) mappings
- not_mapped_to_genome_or_libs: Reads that didn’t map to genome or small RNA libraries
- repetitiveElements: Reads mapping to repetitive regions (LINE, SINE, etc.)
- endogenous_gapped: Reads requiring gapped alignment (spliced reads)
3. Exogenous Alignment (Foreign/Viral Sequences) ⭐
This is the key to your question!
After endogenous alignment, unmapped reads are tested against exogenous (foreign) databases:
- input_to_exogenous_miRNA: Reads sent to exogenous miRNA alignment
- exogenous_miRNA: Reads mapping to viral/foreign miRNAs
- input_to_exogenous_rRNA: Reads sent to exogenous rRNA alignment
- exogenous_rRNA: Reads mapping to foreign rRNA (bacterial, viral)
- input_to_exogenous_genomes: Reads sent to exogenous genome alignment
- exogenous_genomes: Reads mapping to viral/bacterial genomes
🔍 Answering Your Specific Question
For sample nf796:
miRNA_sense: 1.3%
miRNA_antisense: 0.0%
input_to_exogenous_miRNA: 4.4%
exogenous_miRNA: 0.0%Why this pattern?
The pipeline works sequentially:
-
Endogenous miRNA alignment (first pass):
- 1.3% of reads mapped to human endogenous miRNAs (sense strand)
- 0.0% mapped to antisense miRNAs
- These are known human miRNAs from miRBase
-
Exogenous miRNA alignment (second pass):
- The 4.4% represents reads that:
- Did NOT map to endogenous human miRNAs
- Were sent to search viral/foreign miRNA databases
- The 0.0% means none of these 4.4% actually matched viral miRNAs
- These are likely:
- Degraded RNA fragments
- Novel miRNAs not in databases
- Sequencing artifacts
- miRNAs from organisms not in the exogenous database
- The 4.4% represents reads that:
Visual Flow:
reads_used_for_alignment (100%)
↓
[Endogenous alignment]
↓
miRNA_sense (1.3%) ← Human miRNAs
miRNA_antisense (0.0%)
↓
Unmapped reads
↓
[Exogenous alignment]
↓
input_to_exogenous_miRNA (4.4%) ← Sent to viral DB
↓
exogenous_miRNA (0.0%) ← Actually matched viral miRNAs
↓
Remaining 4.4% → discarded or used for other exogenous searchesKey Distinction
| Category | Database | Example |
|---|---|---|
| miRNA_sense | Human miRBase | hsa-miR-21-5p |
| exogenous_miRNA | Viral miRNA DB | EBV-miR-BART1, KSHV-miR-K12 |
Why is input_to_exogenous_miRNA > miRNA_sense?
This is normal and indicates:
- Most reads did NOT map to endogenous miRNAs
- The pipeline casts a wide net, sending many unmapped reads to exogenous searches
- The 4.4% represents “candidate” reads that might be foreign miRNAs
- The 0.0% final match means no viral miRNAs were detected in nf796
If you had a virus-infected sample, you’d see:
input_to_exogenous_miRNA: e.g., 5%exogenous_miRNA: e.g., 3.2% (actual viral miRNA matches)
Other Notable Patterns in Your Heatmap
Looking at your samples:
- nf796, nf797, nf655: Very low genome mapping (~21-45%), high “not_mapped” (~55-79%)
- nf930-nf974: High genome mapping (~66-89%), suggesting better quality or different sample type
- MKL-1 vs WaGa: Different cell lines showing distinct mapping profiles
This suggests nf796/nf797/nf655 might be:
- Lower quality RNA
- Different library preparation
- Higher contamination
- Or genuinely different biology (e.g., more extracellular vesicles with fragmented RNA)
Does this clarify the pipeline stages and the miRNA percentages? Let me know if you need more details about any specific category!