Protected: GEO submission (Ute’s data)
Enter your password to view comments.
Here is the optimized version. Key improvements: (1) it verifies that the two Geneid/gene_name columns from the pasted files are identical and keeps only one copy; (2) it never blindly overwrites column names — R’s automatic header sanitization (check.names=TRUE) produces exactly the names you were assigning by hand (e.g. 042_MKL-1_wt_EV → X042_MKL.1_wt_EV, MKL-1_EV-RNA_87 → MKL.1_EV.RNA_87), so we use those; (3) the final friendly rename is done via an explicit name→name map instead of position-based colnames(x) <- c(...), so a sample can never be silently mislabeled — any mismatch throws an error.
# ---------------------------------------------------------------
# 0) Read the merged count table
# - Do NOT set row.names=1 yet: we first need to compare the two
# Geneid / gene_name columns that come from the two pasted files.
# - check.names=TRUE (default) sanitizes the header deterministically:
# 042_MKL-1_wt_EV -> X042_MKL.1_wt_EV
# MKL-1_EV-RNA_87 -> MKL.1_EV.RNA_87
# 2nd Geneid / gene_name -> Geneid.1 / gene_name.1
# (i.e. exactly the names you previously assigned by hand)
# ---------------------------------------------------------------
d.full <- read.delim2("merged_gene_counts_40samples.txt", sep="\t", header=TRUE)
stopifnot(ncol(d.full) == 44) # 2 x (Geneid + gene_name) + 40 samples
colnames(d.full) # eyeball-check the auto-generated names
# ---------------------------------------------------------------
# 1) Check that the two Geneid / gene_name columns are identical
# If this fails, the two files were pasted in different gene order
# -> stop and fix (diagnosis: which(d.full$Geneid != d.full$Geneid.1))
# ---------------------------------------------------------------
stopifnot(identical(d.full$Geneid, d.full$Geneid.1))
stopifnot(identical(d.full$gene_name, d.full$gene_name.1))
# identical -> keep only one copy of each
d.full$Geneid.1 <- NULL
d.full$gene_name.1 <- NULL
# Ensembl IDs as row names; gene_name was also dropped before DESeq2 previously
stopifnot(!anyDuplicated(d.full$Geneid))
rownames(d.full) <- d.full$Geneid
d.full$Geneid <- NULL
d.full$gene_name <- NULL
stopifnot(ncol(d.full) == 40) # only sample count columns remain
# ---------------------------------------------------------------
# 2) Reorder columns by biological group, using the AUTO-GENERATED names
# (order matches the condition/donor/batch vectors of the old script)
# ---------------------------------------------------------------
col_order <- c(
# MKL-1 RNA
"MKL.1_RNA","MKL.1_RNA_118","MKL.1_RNA_147",
# MKL-1 wt EV
"MKL.1_EV.RNA","MKL.1_EV.RNA_2","MKL.1_EV.RNA_118","MKL.1_EV.RNA_87","MKL.1_EV.RNA_27",
"X042_MKL.1_wt_EV",
# MKL-1 EV DMSO / Dox
"X042_MKL.1_sT_DMSO","X0505_MKL.1_sT_DMSO_EV",
"X042_MKL.1_scr_DMSO_EV","X0505_MKL.1_scr_DMSO_EV",
"X042_MKL.1_sT_Dox","X0505_MKL.1_sT_Dox_EV",
"X042_MKL.1_scr_Dox_EV","X0505_MKL.1_scr_Dox_EV",
# WaGa RNA
"WaGa_RNA","WaGa_RNA_118","WaGa_RNA_147",
# WaGa wt EV
"WaGa_EV.RNA","WaGa_EV.RNA_2","WaGa_EV.RNA_118","WaGa_EV.RNA_147","WaGa_EV.RNA_226",
"X1107_WaGa_wt_EV","X1605_WaGa_wt_EV","X2706_WaGa_wt_EV",
# WaGa EV DMSO / Dox
"X1107_WaGa_sT_DMSO_EV","X1605_WaGa_sT_DMSO_EV","X2706_WaGa_sT_DMSO_EV",
"X1107_WaGa_scr_DMSO_EV","X1605_WaGa_scr_DMSO_EV","X2706_WaGa_scr_DMSO_EV",
"X1107_WaGa_sT_Dox_EV","X1605_WaGa_sT_Dox_EV","X2706_WaGa_sT_Dox_EV",
"X1107_WaGa_scr_Dox_EV","X1605_WaGa_scr_Dox_EV","X2706_WaGa_scr_Dox_EV")
stopifnot(length(col_order) == 40,
!anyDuplicated(col_order),
all(col_order %in% colnames(d.full))) # errors instead of mislabeling
reordered.raw <- d.full[, col_order]
# ---------------------------------------------------------------
# 3) Friendly sample names via an EXPLICIT map (position-independent).
# Kept because all downstream code refers to e.g. "MKL-1 EV sT DMSO 042".
# ---------------------------------------------------------------
name_map <- c(
"MKL.1_RNA" = "MKL-1 RNA",
"MKL.1_RNA_118" = "MKL-1 RNA 118",
"MKL.1_RNA_147" = "MKL-1 RNA 147",
"MKL.1_EV.RNA" = "MKL-1 EV",
"MKL.1_EV.RNA_2" = "MKL-1 EV 2",
"MKL.1_EV.RNA_118" = "MKL-1 EV 118",
"MKL.1_EV.RNA_87" = "MKL-1 EV 87",
"MKL.1_EV.RNA_27" = "MKL-1 EV 27",
"X042_MKL.1_wt_EV" = "MKL-1 EV 042",
"X042_MKL.1_sT_DMSO" = "MKL-1 EV sT DMSO 042",
"X0505_MKL.1_sT_DMSO_EV" = "MKL-1 EV sT DMSO 0505",
"X042_MKL.1_scr_DMSO_EV" = "MKL-1 EV scr DMSO 042",
"X0505_MKL.1_scr_DMSO_EV" = "MKL-1 EV scr DMSO 0505",
"X042_MKL.1_sT_Dox" = "MKL-1 EV sT Dox 042",
"X0505_MKL.1_sT_Dox_EV" = "MKL-1 EV sT Dox 0505",
"X042_MKL.1_scr_Dox_EV" = "MKL-1 EV scr Dox 042",
"X0505_MKL.1_scr_Dox_EV" = "MKL-1 EV scr Dox 0505",
"WaGa_RNA" = "WaGa RNA",
"WaGa_RNA_118" = "WaGa RNA 118",
"WaGa_RNA_147" = "WaGa RNA 147",
"WaGa_EV.RNA" = "WaGa EV",
"WaGa_EV.RNA_2" = "WaGa EV 2",
"WaGa_EV.RNA_118" = "WaGa EV 118",
"WaGa_EV.RNA_147" = "WaGa EV 147",
"WaGa_EV.RNA_226" = "WaGa EV 226",
"X1107_WaGa_wt_EV" = "WaGa EV 1107",
"X1605_WaGa_wt_EV" = "WaGa EV 1605",
"X2706_WaGa_wt_EV" = "WaGa EV 2706",
"X1107_WaGa_sT_DMSO_EV" = "WaGa EV sT DMSO 1107",
"X1605_WaGa_sT_DMSO_EV" = "WaGa EV sT DMSO 1605",
"X2706_WaGa_sT_DMSO_EV" = "WaGa EV sT DMSO 2706",
"X1107_WaGa_scr_DMSO_EV" = "WaGa EV scr DMSO 1107",
"X1605_WaGa_scr_DMSO_EV" = "WaGa EV scr DMSO 1605",
"X2706_WaGa_scr_DMSO_EV" = "WaGa EV scr DMSO 2706",
"X1107_WaGa_sT_Dox_EV" = "WaGa EV sT Dox 1107",
"X1605_WaGa_sT_Dox_EV" = "WaGa EV sT Dox 1605",
"X2706_WaGa_sT_Dox_EV" = "WaGa EV sT Dox 2706",
"X1107_WaGa_scr_Dox_EV" = "WaGa EV scr Dox 1107",
"X1605_WaGa_scr_Dox_EV" = "WaGa EV scr Dox 1605",
"X2706_WaGa_scr_Dox_EV" = "WaGa EV scr Dox 2706")
stopifnot(all(colnames(reordered.raw) %in% names(name_map)),
!anyDuplicated(unname(name_map[colnames(reordered.raw)])))
colnames(reordered.raw) <- unname(name_map[colnames(reordered.raw)])
# ---------------------------------------------------------------
# 4) Write out and filter (same as before)
# ---------------------------------------------------------------
write.csv(reordered.raw, file="counts.txt")
# IMPORTANT: filter low-count genes at this step!
d <- reordered.raw[rowSums(reordered.raw > 3) > 2, ]| Old code | New code | Why |
|---|---|---|
row.names=1 at import |
Import first, compare, then set row names | Allows checking the two Geneid/gene_name columns before one is discarded |
| No check of duplicated id columns | identical() + stopifnot |
Catches a mis-pasted file (different gene order) immediately |
Manual colnames(d.raw) <- c(...43 names...) |
R’s automatic sanitization (which yields the same names) | No forced overwrite; names always come from the file header |
Position-based colnames(reordered.raw) <- c(...) |
Explicit name_map keyed by actual column name |
A sample can never be mislabeled; any unknown/duplicate name throws an error |
Filename "merged_gene_counts_40samples.txt " (trailing space) |
Corrected | The stray space would make read.delim2 fail |
Note: if stopifnot(identical(...)) fails, the two pasted files do not have the same gene order — in that case do not proceed; re-merge them properly (e.g. with a join on Geneid rather than paste/cbind).
Yes — I checked all six metadata vectors against the reordered column order (the 17 MKL-1 + 23 WaGa samples from your col_order/rename step). All vectors have exactly 40 entries, and every entry maps to the correct sample. The code is correct.
| Vector | MKL-1 block (17) | WaGa block (23) | Match? |
|---|---|---|---|
condition_for_pca |
RNA×3, EV×6, sT.DMSO×2, scr.DMSO×2, sT.Dox×2, scr.Dox×2 | RNA×3, EV×8, sT.DMSO×3, scr.DMSO×3, sT.Dox×3, scr.Dox×3 | ✓ |
condition |
MKL1.RNA×3, MKL1.EV×6, then 2+2+2+2 | WaGa.RNA×3, WaGa.EV×8, then 3+3+3+3 | ✓ |
donor |
1,118,147 / 1,2,118,87,27,042 / 042,0505 pairs | 1,118,147 / 1,2,118,147,226,1107,1605,2706 / 1107,1605,2706 ×4 | ✓ |
batch |
2021.08, 2021.09×2 / 2021.08×2, 2021.09×3 / 2022.08×9 | 2021.08, 2021.09×2 / 2021.08×2, 2021.09×3 / 2022.11×15 | ✓ |
cell.line |
MKL-1 ×17 | WaGa ×23 | ✓ |
ids |
unique per sample, order matches | unique per sample, order matches | ✓ |
Spot checks that confirm the alignment:
MKL-1 EV 042 → MKL1.EV, donor 042, batch 2022.08 ✓WaGa EV 1107/1605/2706 → WaGa.EV, donors 1107/1605/2706, batch 2022.11 ✓0505_MKL-1_wt_EV is correctly absent (only one MKL-1 wt EV: 042) ✓stopifnot(ncol(d) == 40,
length(condition) == 40, length(condition_for_pca) == 40,
length(donor) == 40, length(batch) == 40,
length(cell.line) == 40, length(ids) == 40)
# metadata consistent with sample names
stopifnot(all(grepl("^MKL-1", colnames(d)[cell.line == "MKL-1"])),
all(grepl("^WaGa", colnames(d)[cell.line == "WaGa"])))
stopifnot(all(paste0(ifelse(cell.line == "MKL-1", "MKL1", "WaGa"), ".") ==
substr(as.character(condition), 1, 5)))
# visual check — eyeball once before DESeq2
data.frame(sample = colnames(d), condition, donor, batch, cell.line)~batch+condition is full rank here — good. The DMSO/Dox conditions exist only in batch 2022.08 (MKL-1) / 2022.11 (WaGa), but the wt-EV bridge samples (MKL-1 EV 042, WaGa EV 1107/1605/2706) connect those batches to MKL1.EV/WaGa.EV in the earlier batches, so all contrasts remain estimable. DESeq2 will stop with an error if this weren’t the case.donor is recorded but not used in the design. The sT/scr × DMSO/Dox samples are paired by donor (042/0505 and 1107/1605/2706), so a paired design (~batch+donor+condition) could add power — but given your note that you’re unsure whether “rep1” is the same donor across the 2021 and 2022 experiments, keeping ~batch+condition (as in the original analysis) is the safer, consistent choice.condition_for_pca is currently unused (it’s only referenced in a commented-out design line). Harmless, but you can drop it if you want to tidy up.Everything downstream (vst(dds), estimateSizeFactors, the results(dds, name=...) contrast names like MKL1.sT.DMSO_vs_MKL1.scr.DMSO) will work exactly as in your established workflow.
This content is password-protected. To view it, please enter the password below.
https://www.vava8.com/index.php?app=index&act=view&id=122462
由4位牛津大学归国博士创办的津渡生科,自主研发了生命科学垂类大模型 GeneLLM。该模型是全球首个直接基于组学原始数据进行预训练的多组学大模型,相关成果已发表于《Nature Communications》与《Advanced Science》,被视为继AlphaFold(结构预测)和EVO2(基因组理解)之后,向生命”系统级”理解迈进的关键一步。
Token化策略: GeneLLM将约150bp长度的RNA测序片段,通过7碱基滑动窗口(7-mer)切分为”生命Token”,以RNA四种碱基(A/U/G/C)作为基本语义单元,构建生命语言的离散表示。
无监督预训练: 模型采用Transformer架构,在无基因注释、无人工标签的条件下,直接对原始测序数据执行下一碱基预测(Next-Base Prediction)。训练过程处理了约数十万亿条RNA reads,在百卡NVIDIA A100集群上完成,使模型摆脱对已知基因注释的依赖,从原始信号中自主发现潜在生物模式。
两阶段训练流程:
模型规模: 基础版已完成15亿参数、3.5万亿碱基序列预训练;XLarge版本扩展至300亿参数,持续扩大技术壁垒。
多组学覆盖: 训练数据涵盖RNA组、蛋白质组、代谢组等多组学原始数据,具备跨模态生命信息的联合理解能力。
推理效率: 传统方法依赖6Gb深度测序,GeneLLM在1Gb极浅深度测序下仍保持AUC > 0.8,测序成本降低约83%,为大规模临床筛查与普惠精准医疗提供工程可行性。
以GeneLLM为认知底座,津渡生科构建了连接AI与物理实验的完整AI for Science闭环:
认知层——五大智能体协同网络: 文献检索智能体(自动文献综述与假设生成)、实验设计智能体(将实验设计周期从数月压缩至一周)、科学智能体(推理与假设验证)、实验调度智能体(基于通用仪器抽象层统一纳管PCR仪、酶标仪、流式细胞仪、自动化移液工作站等异构设备,内置动态调度算法实现自动排程与冲突规避)、数据分析智能体(结果解读与模型反馈)。
执行层——BioFord Harness: 将实验室转化为可编译、可调度、可观测、可追溯的工程系统,完成三项关键任务:将科学意图或实验DSL编译为跨设备可执行指令;在多设备间完成调度、资源约束管理与异常处理;将实验结果、设备日志和环境参数回流至模型,驱动下一轮迭代。
数据闭环——DBTL循环: 通过Design-Build-Test-Learn闭环,每一次实验(包括失败实验)的参数逻辑、环境记录与错误路径均沉淀为训练数据,形成持续进化的科研数据飞轮。
| 公司/平台 | 路线 | 核心能力 | 当前阶段 |
|---|---|---|---|
| FutureHouse | AI科学大脑 | 文献理解、科学推理 | 数字科研Agent |
| DeepMind | 基础科学模型 | 生物结构预测 | 科学基础模型 |
| Insilico Medicine | AI药物研发 | 靶点发现、分子设计 | AI制药平台 |
| XtalPi晶泰科技 | AI+机器人实验 | 自动化药物研发 | 实验闭环 |
| Lila Sciences | AI Science Factory | 自动化科学工厂 | 重资产实验室 |
| Recursion | 生物数据工业化 | 大规模细胞实验 | 数据驱动研发 |
| Isomorphic Labs | AI药物设计 | AlphaFold路线延伸 | 分子发现 |
| 津渡生科 | 生命大模型+科研Agent | AI理解生命+执行实验 | 全栈AI科研OS |
在这一格局中,津渡生科选择轻量化物理AI路线:不做纯数字AI科学家,不做重资产科学工厂,不做端到端制药管线,而是专注打通模型与实体实验室之间的”最后一公里”基础设施,以实验轨迹、设备接口协议、失败经验库和跨实验室执行网络构建核心护城河。
公司一年内完成4轮融资,投资方包括红杉中国种子基金、创东方投资、南山战新投及高特佳投资(A轮近亿元领投)。
“AI for Science真正的分水岭,不是模型回答得多像科学家,而是实验室能否开始像一个持续学习的系统。”
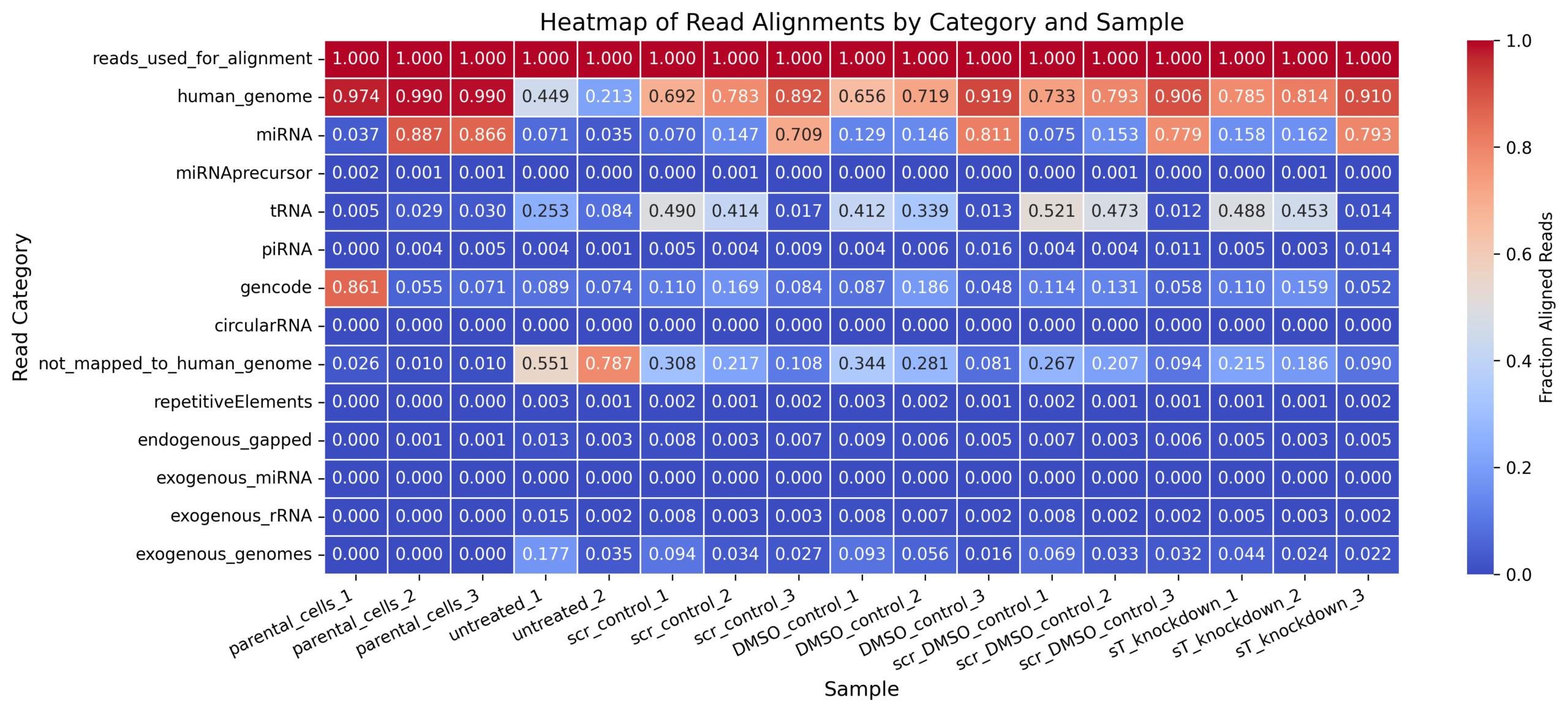
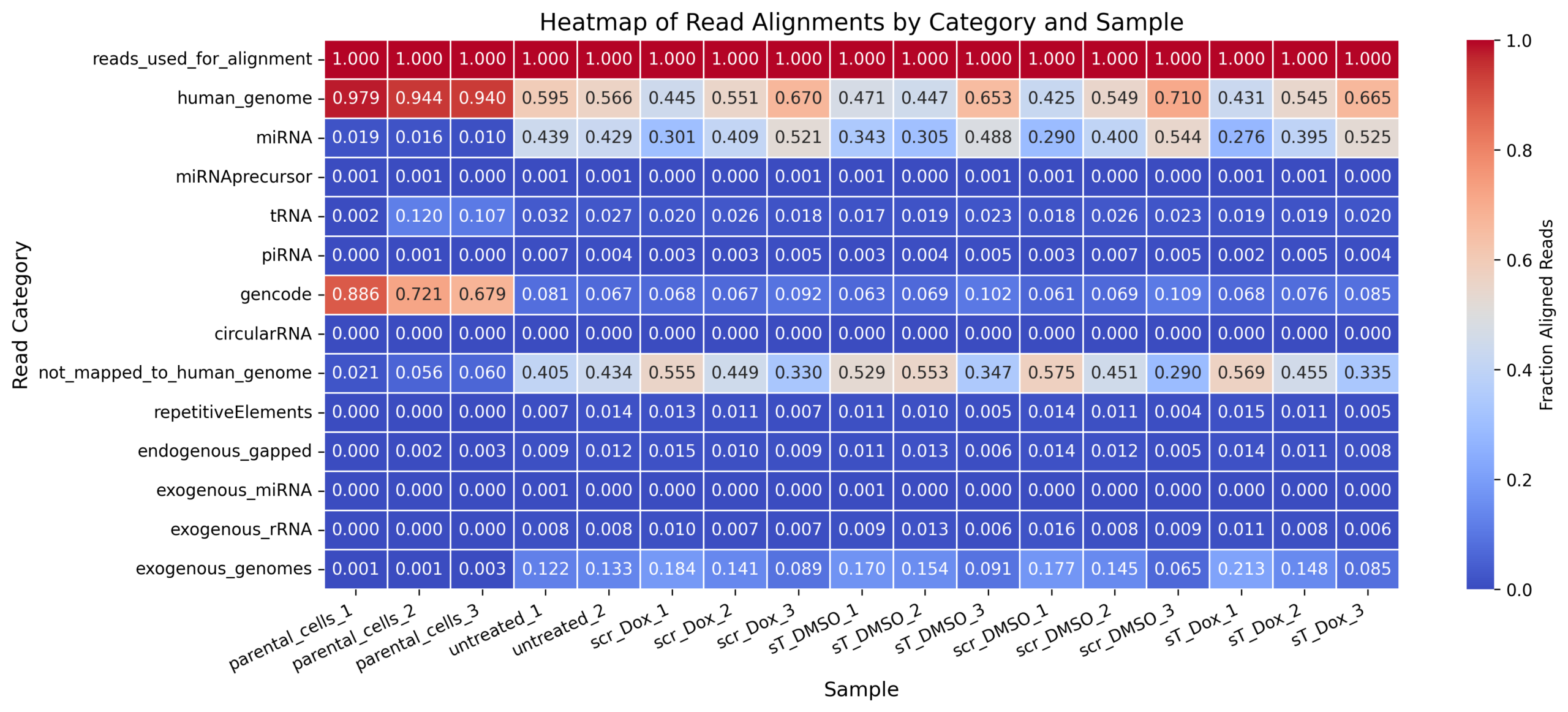
For MKL-1, the two files are complementary, not interchangeable.
exceRpt_biotypeCounts.txt = biotype abundance / composition tableexceRpt_mapping_heatmaps_MKL-1.xlsx = mapping/QC summary table| Feature | exceRpt_biotypeCounts.txt |
exceRpt_mapping_heatmaps_MKL-1.xlsx |
|---|---|---|
| Main purpose | Shows how many reads/abundance estimates were assigned to different RNA biotypes | Shows how reads progressed through QC, trimming, alignment, and mapping categories |
| Data type | Biotype-level abundance table | Mapping/QC fraction table, likely normalized to input reads |
| MKL-1 samples included | Same MKL-1 sample set: 2404_MKL1_wt_EVs, 2608_MKL1_scr_DMSO, 2608_MKL1_scr_Dox, 2608_MKL1_sT_DMSO, 2608_MKL1_sT_Dox, 2608_MKL1_wt_EVs, 2701_MKL1_scr_DMSO, 2701_MKL1_scr_Dox, 2701_MKL1_sT_DMSO, 2701_MKL1_sT_Dox, 2802_MKL1_scr_DMSO, 2802_MKL1_scr_Dox, 2802_MKL1_sT_DMSO, 2802_MKL1_sT_Dox, plus nf780, nf796, nf797 |
Same sample list as the biotypeCounts file |
| Format | Plain text / tab-delimited table | Excel file |
| Values | Numeric abundance values for RNA biotypes. Some values are fractional, suggesting normalized or fractional assignment rather than simple integer raw counts | Values appear to be fractions/proportions, with input = 1 |
| Main biological categories | miRNA, tRNA, piRNA, snRNA, snoRNA, rRNA, protein_coding, lincRNA, retained_intron, processed_transcript, antisense, misc_RNA, exogenous_genomes, exogenous_miRNA, exogenous_rRNA, circularRNA, etc. | input, successfully_clipped, failed_quality_filter, failed_homopolymer_filter, UniVec_contaminants, rRNA, reads_used_for_alignment, genome, miRNA_sense/antisense, tRNA_sense/antisense, piRNA_sense/antisense, gencode_sense/antisense, circularRNA_sense/antisense, not_mapped_to_genome_or_libs, repetitiveElements, exogenous_genomes, etc. |
| Best used for | Small RNA composition plots, e.g. miRNA / tRNA / piRNA / long RNA biotype percentages | Mapping efficiency, QC filtering, alignment statistics, and reproducibility of how reads were distributed |
| Most relevant manuscript panel | Figure 4A and Supplementary Figure S5A: small RNA biotype composition | Mapping/QC text, e.g. percentage of reads mapped, and supplementary QC information |
| Strength | Directly supports the biological small RNA composition results | Directly supports mapping quality and reproducibility |
| Limitation | Does not show QC/mapping steps such as adapter clipping, quality filtering, unmapped reads, etc. | Does not provide the full biotype abundance table needed to reproduce Figure 4A/S5A |
| Repository suitability | High, if converted to clean CSV/TSV and accompanied by sample metadata | Moderate to high, but should be converted from Excel to CSV/TSV and clearly labeled as mapping/QC summary |
| Enough for miRNA-level figures? | No. It gives total miRNA abundance, but not individual miRNA counts/RPM | No. It gives mapping fractions, not individual miRNA abundance |
Submit both, but with different roles:
| File | Submit? | Role |
|---|---|---|
exceRpt_biotypeCounts.txt |
Yes — main processed small RNA biotype file | Supports small RNA biotype composition, e.g. Figure 4A / Suppl. Fig. S5A |
exceRpt_mapping_heatmaps_MKL-1.xlsx |
Yes — secondary mapping/QC file | Supports mapping efficiency and QC reproducibility |
If you can upload only one file, submit:
exceRpt_biotypeCounts.txt
because it is closer to the actual biological small RNA composition results shown in the manuscript.
However, the best processed-data package would be:
smallRNA_exceRpt_biotypeCounts_MKL-1.csv
smallRNA_exceRpt_mapping_summary_MKL-1.csv
smallRNA_sample_metadata_MKL-1.txtConvert the Excel mapping file to CSV/TSV
Many repositories prefer plain text files. For example:
exceRpt_mapping_heatmaps_MKL-1.xlsxcould become:
smallRNA_exceRpt_mapping_summary_MKL-1.csvClarify the value type in exceRpt_biotypeCounts.txt
The values are not all integers. Before submission, check whether this file contains:
Add a short README or column description, for example:
Values are exceRpt-derived biotype abundance estimates.or, if confirmed:
Values are read counts assigned to RNA biotypes by exceRpt.Define the mapping categories
In the mapping heatmap file, categories such as:
reads_used_for_alignmentgenomenot_mapped_to_genome_or_libsgencode_sensemiRNA_senseshould be explained in a README. Otherwise reviewers may not know which row corresponds to “reads mapped to the human genome”.
Decide what to do with nf780, nf796, and nf797
These samples are included in both files but are not MKL-1 EV samples. If they are not part of the manuscript figures, you should either:
You still need an individual miRNA table
Neither of these two files is sufficient for the individual miRNA Manhattan plots or miRNA-level analyses, e.g. Figure 4B or Supplementary Figure S5B. For those, you should also provide:
smallRNA_miRNA_counts_all_samples.txt
smallRNA_miRNA_RPM_normalized_all_samples.txtif those were used for the figures.
For MKL-1:
Primary processed file to submit:
exceRpt_biotypeCounts.txt
→ rename to smallRNA_exceRpt_biotypeCounts_MKL-1.csv
Secondary processed file to submit:
exceRpt_mapping_heatmaps_MKL-1.xlsx
→ convert to smallRNA_exceRpt_mapping_summary_MKL-1.csvIf only one file can be submitted, choose exceRpt_biotypeCounts.txt.
If you want full reproducibility, submit both, plus a sample metadata file.
Yes — but you do not necessarily need to submit every intermediate pipeline file.
You should submit the final processed files that are required to reproduce the quantitative results/figures in the manuscript.
By “raw sequencing data” I assume you mean FASTQ/BAM. Files such as *_raw_counts*.txt are already processed data relative to FASTQ, and they are usually expected for GEO/journal submission if they underlie the figures.
Below is a careful breakdown based on the manuscript text.
| # | Processed data type | Where it is used in the manuscript | Original manuscript sentence / relevant text | Suggested file(s) to submit |
|---|---|---|---|---|
| 1 | RNA-seq gene-level count matrix | Methods 4.10; Results on EV RNA cargo; Fig. 3; Suppl. Fig. S4, S9, S11 | “A total of 40 RNA-seq libraries were processed using the nf-core/rnaseq pipeline…” “Gene-level read counts were generated using featureCounts…” “Raw count data were analyzed using DESeq2.” |
RNAseq_raw_counts_all_samples.txt |
| 2 | RNA-seq normalized / VST-transformed counts | Methods 4.10; heatmaps/PCA/clustering; Fig. 6D; Suppl. Fig. S9 | “For visualization, count data were normalized and variance-stabilized using the variance stabilizing transformation (VST).” | RNAseq_VST_normalized_counts_all_samples.txt or similar |
| 3 | RNA-seq differential abundance tables | Fig. 3B; Fig. 6D; Suppl. Fig. S9; Results on EV vs parental cells and sT knockdown | “Approximately 26,000 transcripts showed a higher relative abundance in EVs, whereas approximately 2,800 transcripts exhibited a lower relative abundance compared with the parental cells.” “Fifteen transcripts showed significantly higher relative abundance following sT knockdown…” |
RNAseq_DESeq2_EV_vs_parental_results.txt RNAseq_DESeq2_sT_knockdown_results.txt |
| 4 | Viral MCPyV transcript counts / normalized abundance | Methods 4.10; Suppl. Fig. S11 | “Sequencing reads were aligned using STAR against a combined reference comprising the human genome (GRCh38) and the corresponding Merkel cell polyomavirus (MCPyV) genome…” Suppl. Fig. S11: “Red dots indicate MCPyV-derived viral transcripts…” |
RNAseq_MCPyV_viral_transcript_counts.txt or include viral rows in the main RNA-seq count table |
| 5 | small RNA-seq miRNA count matrix | Methods 4.11; Fig. 4; Fig. 5; Suppl. Fig. S5, S7 | “Known human miRNAs were annotated according to miRBase, and read counts for individual miRNAs were generated using the COMPSRA pipeline.” “Raw miRNA count data were analyzed using DESeq2.” |
smallRNA_miRNA_counts_all_samples.txt |
| 6 | small RNA-seq normalized miRNA abundance | Fig. 4B; Suppl. Fig. S5B; possibly S7 | Fig. 4B legend: “Manhattan plots showing normalized miRNA abundance(log10 reads per million; RPM)…” | smallRNA_miRNA_RPM_normalized_all_samples.txt or VST/DESeq2-normalized miRNA table |
| 7 | small RNA biotype composition table | Fig. 4A; Suppl. Fig. S5A; Results on small RNA composition | “In parental cells, 59% of mapped reads were annotated as miRNAs…” “In WaGa-derived EVs, 28% of mapped reads were annotated as miRNAs… tRNA-derived reads increased to approximately 29% and piRNA-derived reads accounted for 0.65%.” |
smallRNA_biotype_counts_summary.txt or the source count table used to generate Fig. 4A/S5A |
| 8 | small RNA mapping summary | Results on small RNA mapping; Fig. 4A context | “Approximately 98% of reads obtained from WaGa cells mapped to the human genome, whereas approximately 73% of reads from EVs could be mapped to the human genome.” | smallRNA_mapping_summary.txt |
| 9 | RBP motif enrichment results | Results on RNA-binding protein motifs; Fig. 3 / Fig. S4 | “Analysis of Motif Enrichment(AME) was performed using the ATtRACT database.” “Several significantly enriched sequence motifs and their corresponding RBPs were identified.” |
RNAseq_RBP_motif_enrichment_results.txt |
| 10 | miRNA target network table | Results 2.5; Fig. 5; Suppl. Fig. S6 | “…experimentally validated target genes of the 15 most abundant miRNAs identified in WaGa-derived EVs were retrieved from miRTarBase and used to construct a miRNA-target interaction network.” “The resulting network comprised 196 target genes.” |
miRNA_target_network_nodes.txt miRNA_target_network_edges.txt |
| 11 | Proteomics protein identification and quantification table | Results proteomics; Fig. 2; Fig. 6; Suppl. Fig. S3, S10; Data Availability | “To characterize the protein cargo of WaGa-derived EVs, mass spectrometry was performed, identifying 608 proteins consistently detected across all biological replicates.” “The proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository…” |
Proteomics_protein_identifications_quantification.txt optionally peptide-level table |
| 12 | Proteomics differential abundance table | Fig. 6C; Suppl. Fig. S10; Results on sT knockdown proteome | “Differential abundance analysis identified 26 proteins whose abundance was significantly altered following sT knockdown, comprising 24 proteins with higher abundance and 2 proteins with lower abundance in EVs.” | Proteomics_DE Proteins_sT_knockdown_WaGa.txt Proteomics_DE Proteins_sT_knockdown_MKL-1.txt |
| File | My recommendation | Reason |
|---|---|---|
RNAseq_raw_counts_all_samples.txt |
Yes — submit | This is the count matrix used for DESeq2 analysis and underlies Fig. 3, Fig. 6D, Suppl. Fig. S9, and likely Suppl. Fig. S11. The manuscript explicitly says: “Raw count data were analyzed using DESeq2.” |
smallRNA_miRNA_counts_all_samples.txt |
Yes — submit | This is the main small RNA count matrix used for miRNA abundance, differential miRNA analysis, Fig. 4, Fig. 5, Suppl. Fig. S5 and S7. The manuscript explicitly says miRNA read counts were generated and analyzed with DESeq2. |
smallRNA_gencode_counts_all_samples.txt |
Conditional | Submit only if this file was used to generate the small RNA biotype composition shown in Fig. 4A / Suppl. Fig. S5A, e.g. miRNA, tRNA, piRNA, long RNA, etc. The manuscript does not explicitly mention “GENCODE counts”, so it is not automatically required. If this file is only an intermediate file and the biotype percentages were generated from another summary table, you can submit the summary table instead. |
smallRNA_tRNA_counts_all_samples.txt |
Conditional / optional | The manuscript only reports tRNAs as a class: “tRNAs represented 29% of mapped reads…” It does not appear to analyze individual tRNA-derived fragments in detail. If this file is needed to calculate the tRNA percentage in Fig. 4A/S5A, submit it or include it in a combined biotype table. If not, a summarized biotype count table is enough. |
smallRNA_piRNA_counts_all_samples.txt |
Conditional / optional | Same logic as tRNA. The manuscript only reports piRNAs as a percentage: “piRNAs accounted for 0.65%.” If this file is required to reproduce that number, submit it or include it in a biotype summary. Otherwise, individual piRNA counts are not strictly needed unless they are analyzed elsewhere. |
smallRNA_mapping_summary.txt |
Recommended, but not always mandatory | This supports the mapping rates and possibly the biotype composition reported in the manuscript: “Approximately 98% of reads… mapped… whereas approximately 73%…” It is very useful for reproducibility. I would include it as a supplementary processed file or as part of a small RNA QC/summary table. |
I would submit the following as the clean, final processed files:
| File | Needed? | Why |
|---|---|---|
RNAseq_raw_counts_all_samples.txt |
Yes | Underlies DESeq2 analysis |
RNAseq_normalized_counts_all_samples.txt |
Strongly recommended | The manuscript says VST-normalized data were used for visualization |
RNAseq_DESeq2_results_EV_vs_parental.txt |
Yes | Underlies volcano plots / differential RNA cargo |
RNAseq_DESeq2_results_sT_knockdown.txt |
Yes | Underlies Fig. 6D / Suppl. Fig. S9 |
RNAseq_MCPyV_transcript_counts_or_RPM.txt |
Recommended | Needed if Suppl. Fig. S11 is shown |
RNAseq_sample_metadata.txt |
Yes | Needed to understand conditions, replicates, EV vs cells, Dox/DMSO, WaGa/MKL-1 |
| File | Needed? | Why |
|---|---|---|
smallRNA_miRNA_counts_all_samples.txt |
Yes | Main miRNA count matrix |
smallRNA_miRNA_RPM_normalized_all_samples.txt |
Strongly recommended | Fig. 4B and Suppl. Fig. S5B show normalized miRNA abundance in RPM |
smallRNA_biotype_counts_summary.txt |
Yes / strongly recommended | Underlies Fig. 4A and Suppl. Fig. S5A |
smallRNA_mapping_summary.txt |
Recommended | Supports mapping rates reported in the text |
smallRNA_DESeq2_miRNA_results.txt |
Yes if differential miRNA analysis is shown | Suppl. Fig. S7 / related text |
smallRNA_sample_metadata.txt |
Yes | Needed to interpret samples |
If you create one clean file called, for example:
smallRNA_biotype_counts_summary.txtwith columns like:
sample
cell_line
sample_type
treatment
total_reads
mapped_reads
miRNA_counts
miRNA_percent
tRNA_counts
tRNA_percent
piRNA_counts
piRNA_percent
longRNA_counts
longRNA_percent
other_counts
other_percentthen you may not need to submit separate smallRNA_tRNA_counts_all_samples.txt and smallRNA_piRNA_counts_all_samples.txt, unless those files are the direct source of the figure.
The manuscript says:
“The proteomics data have been deposited with the ProteomeXchange Consortium via the PRIDE partner repository under accession number PXDXXXXX.”
And your manuscript notes also say:
“Bente has to do this: Upload processed files (e.g., peptide/protein identifications, quantification tables). Include metadata describing the experimental design.”
So for proteomics, you should submit at least:
| Proteomics processed file | Needed? |
|---|---|
| Protein identification table | Yes |
| Protein quantification table | Yes |
| Peptide identification table | Recommended / often expected by PRIDE |
| Differential protein abundance table for sT knockdown | Yes |
| Sample metadata for MS runs | Yes |
You should also check these points, because they affect which processed files are correct to submit.
In the Results section, the manuscript says:
“Sequencing data were analyzed using the exceRpt pipeline, which is optimized for extracellular RNA analysis.”
But in Methods 4.11, the manuscript says:
“Raw FASTQ files generated by small RNA sequencing were processed using Cutadapt…”
“High-quality reads were aligned… using COMPSRA with the STAR aligner.”
You need to decide which pipeline actually produced the final count files:
This matters because the submitted processed files must match the described pipeline.
Methods 4.11 says:
“…combined reference comprising the human genome (GRCh38) and the Merkel cell polyomavirus genome(JN707599)…”
But for RNA-seq, Methods 4.10 says:
“…combined reference comprising the human genome(GRCh38) and the corresponding Merkel cell polyomavirus(MCPyV) genome(KJ128379.1 for WaGa samples and FJ173815.1 for MKL-1 samples).”
Check whether small RNA-seq really used only JN707599 for all samples, or whether it should match the WaGa/MKL-1 references.
In the Results text, Figure 3C and 3D may not match the Figure legend perfectly. The Results text describes GO enrichment and then RBP motif enrichment, while the Figure legend appears to assign them differently. Before submission, make sure the text, figure panels, and supplementary files match.
For your listed files:
RNAseq_raw_counts_all_samples.txt → Yes, submit
smallRNA_miRNA_counts_all_samples.txt → Yes, submit
smallRNA_gencode_counts_all_samples.txt → Only if used to generate the reported small RNA biotype composition
smallRNA_tRNA_counts_all_samples.txt → Optional unless needed to reproduce Fig. 4A/S5A
smallRNA_piRNA_counts_all_samples.txt → Optional unless needed to reproduce Fig. 4A/S5A
smallRNA_mapping_summary.txt → Recommended, useful for reproducibilityThe safest approach is to submit a clean final processed-data package containing:
Yes, absolutely. Based on the manuscript’s Data Availability Statement and the email from Nicole Fischer:
| Data Type | Repository | Accession Format |
|---|---|---|
| RNA-seq (total RNA) | NCBI GEO (linked to SRA) | GSEXXXXX / SRXXXXX |
| Small RNA-seq | NCBI GEO (linked to SRA) | GSEXXXXX / SRXXXXX |
| Proteomics (LC-MS/MS) | PRIDE / ProteomeXchange | PXDXXXXX |
.fastq.gz files → uploaded to SRA (Sequence Read Archive) via GEO⚠️ Critical: The email says data must be uploaded but NOT released publicly yet. In GEO you set a future release date (e.g., 1–2 years from now) so you get the accession number immediately but data stays private until manuscript publication.
本研究生成的RNA测序(RNA-seq)和小RNA测序(small RNA-seq)数据集已存入基因表达综合数据库(GEO),登录号为GSEXXXXX。蛋白质组学数据已存入ProteomeXchange联盟的PRIDE合作存储库,登录号为PXDXXXXX。
Before you start, gather:
| Item | Details |
|---|---|
| NCBI Account | Register at ncbi.nlm.nih.gov/account |
| GEO Account | After NCBI login, request GEO submission access at geo@ncbi.nlm.nih.gov |
| Raw FASTQ files | All .fastq.gz files (RNA-seq + small RNA-seq) |
| Processed data | Count matrices (featureCounts output for RNA-seq; COMPSRA miRNA counts for small RNA-seq) |
| Sample metadata | Cell line, condition, replicate, sample type (EV vs parental cell) |
| Protocol info | Library prep kits, sequencing platform, read length |
Based on the manuscript, organize samples into a clear table. Here is the suggested sample organization:
| Sample Name | Cell Line | Sample Type | Condition | Replicate | File |
|---|---|---|---|---|---|
| WaGa_wt_EV_rep1 | WaGa | EV | untreated (wt) | 1 | 1107_WaGa_wt_EV.fastq.gz |
| WaGa_wt_EV_rep2 | WaGa | EV | untreated (wt) | 2 | 1605_WaGa_wt_EV.fastq.gz |
| WaGa_wt_EV_rep3 | WaGa | EV | untreated (wt) | 3 | 2706_WaGa_wt_EV.fastq.gz |
| WaGa_scr_DMSO_EV_rep1 | WaGa | EV | scr + DMSO | 1 | 1107_WaGa_scr_DMSO_EV.fastq.gz |
| WaGa_scr_Dox_EV_rep1 | WaGa | EV | scr + Dox | 1 | 1107_WaGa_scr_Dox_EV.fastq.gz |
| WaGa_sT_DMSO_EV_rep1 | WaGa | EV | sT + DMSO | 1 | 1107_WaGa_sT_DMSO_EV.fastq.gz |
| WaGa_sT_Dox_EV_rep1 | WaGa | EV | sT + Dox (sT KD) | 1 | 1107_WaGa_sT_Dox_EV.fastq.gz |
| … (rep2, rep3) | |||||
| WaGa_cell_RNA_rep1 | WaGa | Parental cell | untreated | 1 | WaGa_RNA.fastq.gz |
| WaGa_cell_RNA_rep2 | WaGa | Parental cell | untreated | 2 | WaGa_RNA_118.fastq.gz |
| WaGa_cell_RNA_rep3 | WaGa | Parental cell | untreated | 3 | WaGa_RNA_147.fastq.gz |
Same structure as WaGa.
Map nf774, nf930–nf939, nf961, nf962, nf971–nf974 to their corresponding conditions. You need to check your lab records to confirm which nf-number corresponds to which sample.
Map 2404_MKL1_wt_EVs, 2608_*, 2701_*, 2802_*, nf780, nf796, nf797 similarly.
⚠️ Important: The file naming is inconsistent (some have EV suffix, some don’t; some use dates like 042/0505, others use nf-numbers). You must verify the sample-to-file mapping with Ute or your lab notebook before submission.
| Field | What to Enter |
|---|---|
| Title | Multi-omics characterization of extracellular vesicles derived from virus-positive Merkel cell carcinoma cells – RNA-seq and small RNA-seq |
| Summary | Copy/adapt from manuscript abstract |
| Overall design | Two MCPyV-positive MCC cell lines (WaGa and MKL-1) with doxycycline-inducible sT knockdown. EVs isolated by differential ultracentrifugation. Total RNA-seq and small RNA-seq of EVs and parental cells under sT knockdown and control conditions. Three biological replicates per condition. |
| Experiment type | RNA-Seq; small RNA-seq |
| Release date | Set to a future date (e.g., 2027-08-01 or later) to keep data private |
For each sample, provide:
| Field | Value |
|---|---|
| Instrument | Illumina NextSeq 500 |
| Read length | 75 bp single-end |
| Library strategy | RNA-Seq / miRNA-Seq |
| Library source | transcriptomic |
| Library selection | cDNA / size fractionation (small RNA) |
| Library kit | CORALL Total RNA-Seq V2 Kit / LEXOGEN Small RNA-Seq Library Prep Kit |
ftp://ftp-private.ncbi.nlm.nih.gov/uploads/geo/...).fastq.gz files:# Example using FTP (you can also use Aspera or web browser)
# Connect to the FTP server provided by GEO
ftp ftp-private.ncbi.nlm.nih.gov
# Login with provided credentials
cd uploads/geo/your_folder/
# Upload WaGa RNA-seq
put Data_RNA-Seq_WaGa/1107_WaGa_wt_EV.fastq.gz
put Data_RNA-Seq_WaGa/1107_WaGa_scr_DMSO_EV.fastq.gz
# ... repeat for all files
# Upload small RNA-seq
put Data_smallRNA_WaGa/nf774.fastq.gz
# ... repeat for all filesOr use Aspera (faster for large files):
ascp -k 1 -T -l 300m \
Data_RNA-Seq_WaGa/*.fastq.gz \
subasp@upload.ncbi.nlm.nih.gov:uploads/geo/your_folder/RNASeq_WaGa/
ascp -k 1 -T -l 300m \
Data_smallRNA_WaGa/*.fastq.gz \
subasp@upload.ncbi.nlm.nih.gov:uploads/geo/your_folder/smallRNA_WaGa/GEO requires processed data. Prepare:
RNA-seq count matrix (from featureCounts):
Small RNA-seq miRNA count matrix (from COMPSRA):
Upload these as supplementary files in the GEO submission portal:
.txt or .csv (tab-delimited)WaGa_MKL1_RNAseq_raw_counts.txt, WaGa_MKL1_smallRNA_miRNA_counts.txtReplace GSEXXXXX in the Data Availability Statement with the actual GSE number:
“The RNA sequencing (RNA-seq) and small RNA sequencing (small RNA-seq) datasets generated in this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE[ACTUAL NUMBER].”
| Step | Action | Status |
|---|---|---|
| ☐ 1 | Register NCBI + GEO account | |
| ☐ 2 | Verify sample-to-file mapping (especially nf-numbers!) | |
| ☐ 3 | Prepare metadata spreadsheet | |
| ☐ 4 | Generate processed count matrices | |
| ☐ 5 | Start GEO submission (High-Throughput Sequencing) | |
| ☐ 6 | Fill in Series, BioSample, SRA metadata | |
| ☐ 7 | Upload FASTQ files via FTP/Aspera | |
| ☐ 8 | Upload processed data files | |
| ☐ 9 | Set future release date (NOT public) | |
| ☐ 10 | Submit → receive GSE accession number | |
| ☐ 11 | Send GSE number to Nicole/Ute for manuscript |
💡 Tip: The proteomics data (PRIDE/ProteomeXchange) is a separate submission handled by the mass spectrometry team (Bente Siebels). You only need to handle the RNA-seq and small RNA-seq GEO submission.
This content is password-protected. To view it, please enter the password below.
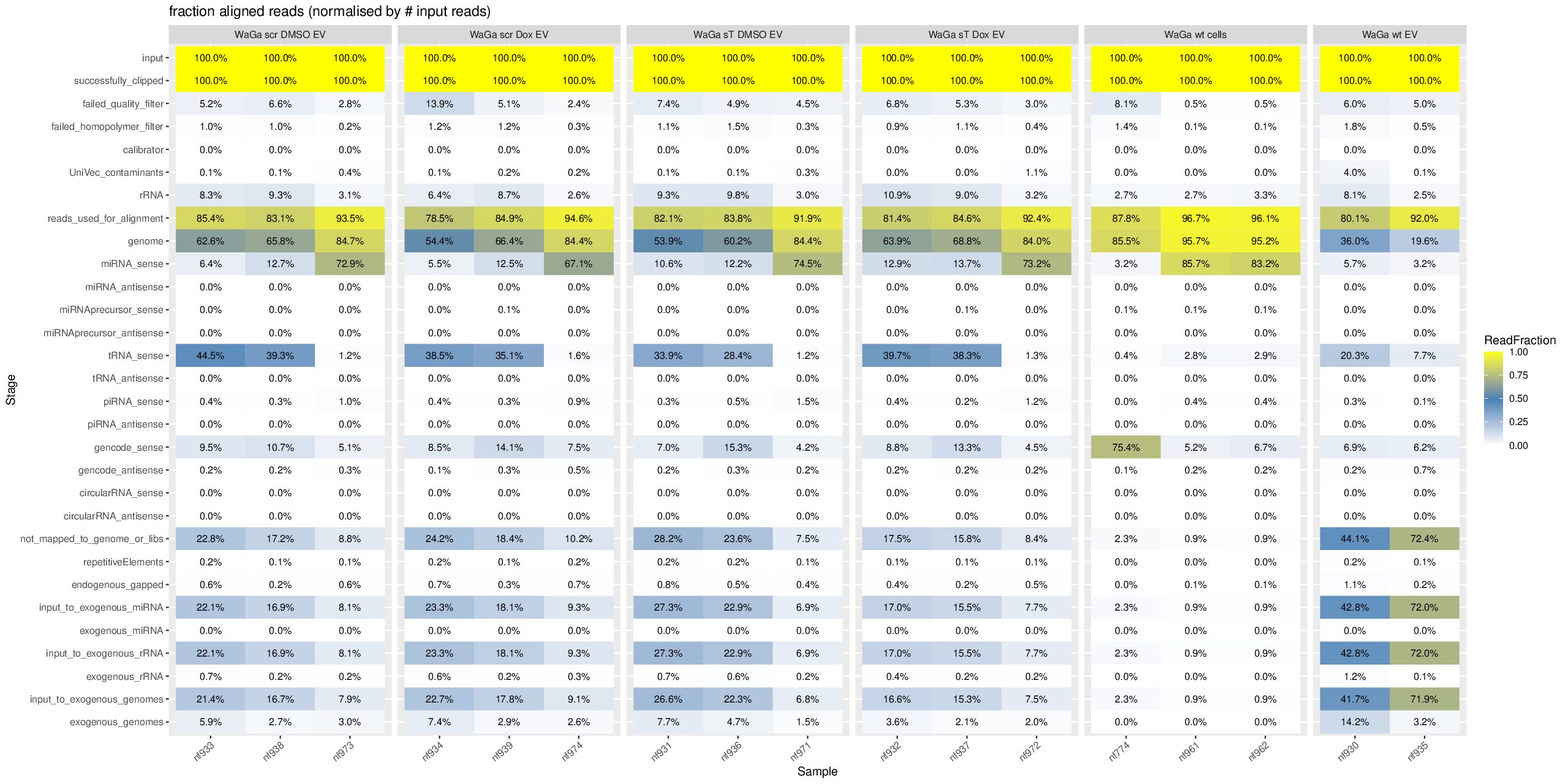
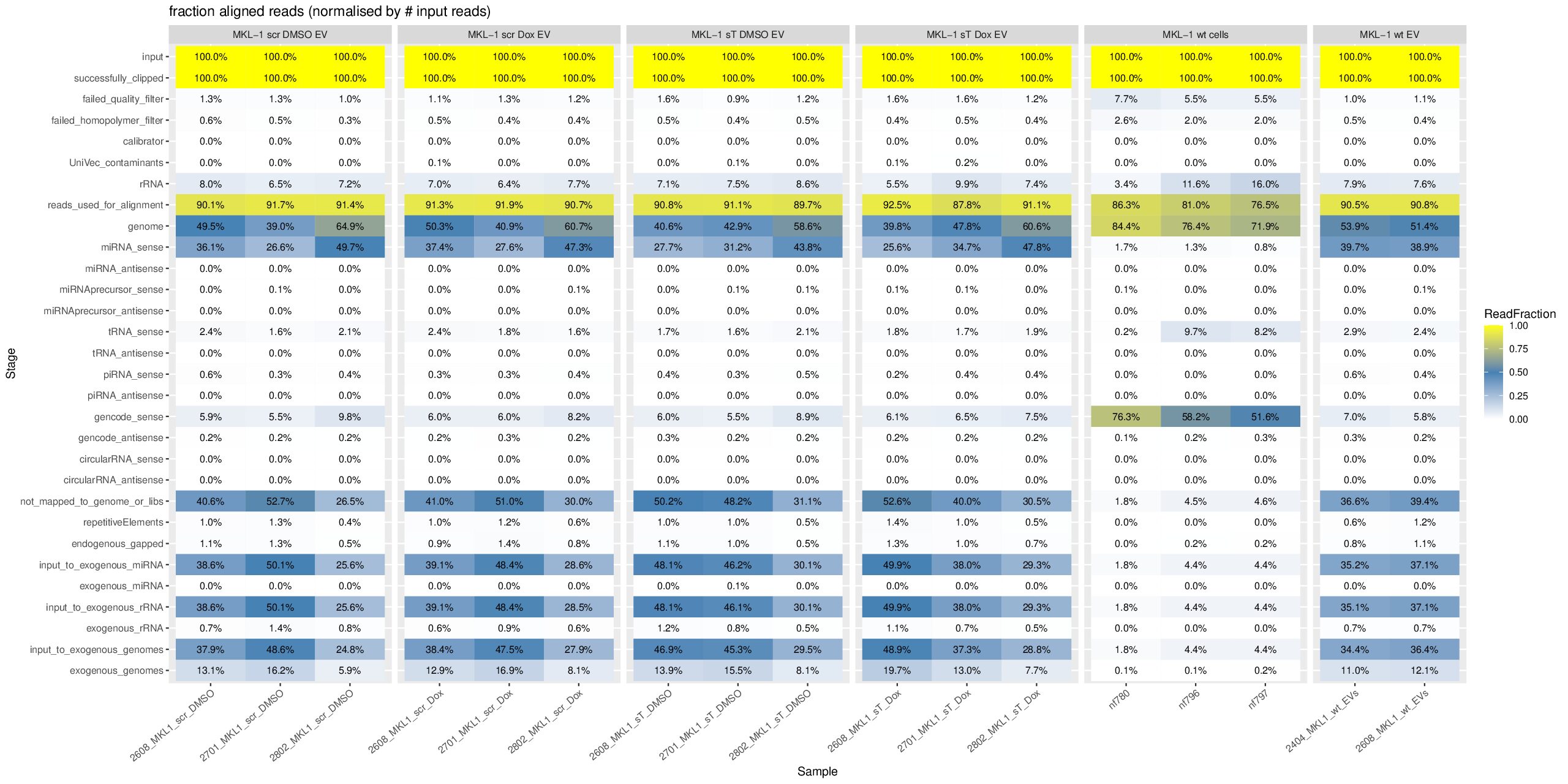
Regarding your question why the miRNA percentage of the first sample, nf774, in the group “WaGa wt cells” is totally different from the other two samples, nf961 and nf962, which show 85.7% and 83.2% miRNA in exceRpt_DiagnosticPlots_WaGa.pdf, I think the drastic difference is due to a batch effect.
More specifically, nf774 was generated in an older sequencing/library preparation batch, whereas nf961 and nf962 were generated in a later optimized batch. Between these batches, the small RNA enrichment/size-selection strategy was different. The older nf774 library likely had less efficient small RNA enrichment and/or stronger adapter/junk read contamination, while nf961 and nf962 benefited from the optimized protocol and therefore show high miRNA percentages. Thus, this difference is most likely technical rather than biological.
Please find the updated sample summary and batch information below.
| Sample ID | Cell Line / Type (PDF Label) | Project Batch (Sequencing Run ID) |
|---|---|---|
| Wild-Type Cells | ||
| nf961 | WaGa wt cells | 250411_VH00358_135_AAGKGLHM5 |
| nf962 | WaGa wt cells | 250411_VH00358_135_AAGKGLHM5 |
| nf774 | WaGa wt cells | 220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti |
| nf780 | MKL-1 wt cells | 220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti |
| nf796 | MKL-1 wt cells | 221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti |
| nf797 | MKL-1 wt cells | 221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti |
| WaGa EV Samples | ||
| nf657 (Excluded) | WaGa wt EV | 210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti |
| nf930, nf935 | WaGa wt EV | 231016_NB501882_0435_AHG7HMBGXV |
| nf931, nf936 | WaGa sT DMSO EV | 231016_NB501882_0435_AHG7HMBGXV |
| nf971 | WaGa sT DMSO EV | 250411_VH00358_135_AAGKGLHM5 |
| nf932, nf937 | WaGa sT Dox EV | 231016_NB501882_0435_AHG7HMBGXV |
| nf972 | WaGa sT Dox EV | 250411_VH00358_135_AAGKGLHM5 |
| nf933, nf938 | WaGa scr DMSO EV | 231016_NB501882_0435_AHG7HMBGXV |
| nf973 | WaGa scr DMSO EV | 250411_VH00358_135_AAGKGLHM5 |
| nf934, nf939 | WaGa scr Dox EV | 231016_NB501882_0435_AHG7HMBGXV |
| nf974 | WaGa scr Dox EV | 250411_VH00358_135_AAGKGLHM5 |
| MKL-1 EV Samples | ||
| nf655 (Excluded) | MKL-1 wt EV | 210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti |
| 2404, 2608 | MKL-1 wt EV | 20260506_AV243904_0073_A |
| 2608, 2701, 2802 | MKL-1 sT DMSO EV | 20260506_AV243904_0073_A |
| 2608, 2701, 2802 | MKL-1 sT Dox EV | 20260506_AV243904_0073_A |
| 2608, 2701, 2802 | MKL-1 scr DMSO EV | 20260506_AV243904_0073_A |
| 2608, 2701, 2802 | MKL-1 scr Dox EV | 20260506_AV243904_0073_A |
The 6 wild-type cell samples originate from 3 distinct sequencing runs:
220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti — June 2022
221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti — December 2022
250411_VH00358_135_AAGKGLHM5 — April 2025
In summary, nf774 comes from a different and earlier batch than nf961/nf962, which supports the interpretation that the large difference in miRNA percentage is mainly caused by batch effects and differences in library preparation/small RNA enrichment strategy.