TODO: 你能不能再帮我准备一个 Manhattan 图:一列是 WaGa 细胞,一列是未处理的 WaGa EVs,其中每一列显示的是所有重复实验的平均 read count(细胞一列、EVs 一列)?另外,你可以把丰度最高的 20 个 miRNA 标注出来吗?这个生成起来需要很久吗? 另外有个小提醒,可能 Carmen(我们组的临床科学家)周一也会需要类似的图,不过标签会不一样。
manhattan_plot_top_miRNAs_based_on_mean_RPM.R
# see http://xgenes.com/article/article-content/288/draw-plots-for-mirnas-generated-by-compsra/
# see http://xgenes.com/article/article-content/289/draw-plots-for-pirna-generated-by-compsra/
# see http://xgenes.com/article/article-content/290/draw-plots-for-snrna-generated-by-compsra/
#Input file
#exceRpt_miRNA_ReadCounts.txt
#exceRpt_piRNA_ReadCounts.txt
cd ~/DATA/Data_Ute/Data_Ute_smallRNA_7/summaries_exo7
mamba activate r_env
R
#> .libPaths()
#[1] "/home/jhuang/mambaforge/envs/r_env/lib/R/library"
#BiocManager::install("AnnotationDbi")
#BiocManager::install("clusterProfiler")
#BiocManager::install(c("ReactomePA","org.Hs.eg.db"))
#BiocManager::install("limma")
#BiocManager::install("sva")
#install.packages("writexl")
#install.packages("openxlsx")
library("AnnotationDbi")
library("clusterProfiler")
library("ReactomePA")
library("org.Hs.eg.db")
library(DESeq2)
library(gplots)
library(limma)
library(sva)
#library(writexl) #d.raw_with_rownames <- cbind(RowNames = rownames(d.raw), d.raw); write_xlsx(d.raw, path = "d_raw.xlsx");
library(openxlsx)
setwd("../summaries_exo7/")
d.raw<- read.delim2("exceRpt_miRNA_ReadCounts.txt",sep="\t", header=TRUE, row.names=1)
# Desired column order
desired_order <- c(
"parental_cells_1", "parental_cells_2", "parental_cells_3",
"untreated_1", "untreated_2",
"scr_control_1", "scr_control_2", "scr_control_3",
"DMSO_control_1", "DMSO_control_2", "DMSO_control_3",
"scr_DMSO_control_1", "scr_DMSO_control_2", "scr_DMSO_control_3",
"sT_knockdown_1", "sT_knockdown_2", "sT_knockdown_3"
)
# Reorder columns
d.raw <- d.raw[, desired_order]
setdiff(desired_order, colnames(d.raw)) # Shows missing or misnamed columns
#sapply(d.raw, is.numeric)
d.raw[] <- lapply(d.raw, as.numeric)
#d.raw[] <- lapply(d.raw, function(x) as.numeric(as.character(x)))
d.raw <- round(d.raw)
write.csv(d.raw, file ="d_raw.csv")
write.xlsx(d.raw, file = "d_raw.xlsx", rowNames = TRUE)
# ------ Code sent to Ute ------
#d.raw <- read.delim2("d_raw.csv",sep=",", header=TRUE, row.names=1)
parental_or_EV = as.factor(c("parental","parental","parental", "EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV"))
#donor = as.factor(c("0505","1905", "0505","1905", "0505","1905", "0505","1905", "0505","1905", "0505","1905"))
batch = as.factor(c("Aug22","March25","March25", "Sep23","Sep23", "Sep23","Sep23","March25", "Sep23","Sep23","March25", "Sep23","Sep23","March25", "Sep23","Sep23","March25"))
replicates = as.factor(c("parental_cells","parental_cells","parental_cells", "untreated","untreated", "scr_control","scr_control","scr_control", "DMSO_control","DMSO_control","DMSO_control", "scr_DMSO_control", "scr_DMSO_control","scr_DMSO_control", "sT_knockdown", "sT_knockdown", "sT_knockdown"))
ids = as.factor(c("parental_cells_1", "parental_cells_2", "parental_cells_3",
"untreated_1", "untreated_2",
"scr_control_1", "scr_control_2", "scr_control_3",
"DMSO_control_1", "DMSO_control_2", "DMSO_control_3",
"scr_DMSO_control_1", "scr_DMSO_control_2", "scr_DMSO_control_3",
"sT_knockdown_1", "sT_knockdown_2", "sT_knockdown_3"))
cData = data.frame(row.names=colnames(d.raw), replicates=replicates, ids=ids, batch=batch, parental_or_EV=parental_or_EV)
dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates+batch)
# Filter low-count miRNAs
dds <- dds[ rowSums(counts(dds)) > 10, ] #1322-->903
rld <- rlogTransformation(dds)
# ----------- manhattan_plot -------------
# Load the required libraries
library(ggplot2)
library(dplyr)
library(tidyr)
library(ggrepel) # For better label positioning
# Step 1: Compute RPM from raw counts (d.raw has miRNAs in rows, samples in columns)
d.raw_5 <- d.raw[, 1:5] # assuming 5 samples
total_counts <- colSums(d.raw_5)
RPM <- sweep(d.raw_5, 2, total_counts, FUN = "/") * 1e6
# Step 2: Prepare long-format dataframe
RPM$miRNA <- rownames(RPM)
df <- pivot_longer(RPM, cols = -miRNA, names_to = "sample", values_to = "RPM")
# Step 3: Log-transform RPM
df <- df %>%
mutate(logRPM = log10(RPM + 1))
# Step 4: Add miRNA index for x-axis positioning
df <- df %>%
arrange(miRNA) %>%
group_by(sample) %>%
mutate(Position = row_number())
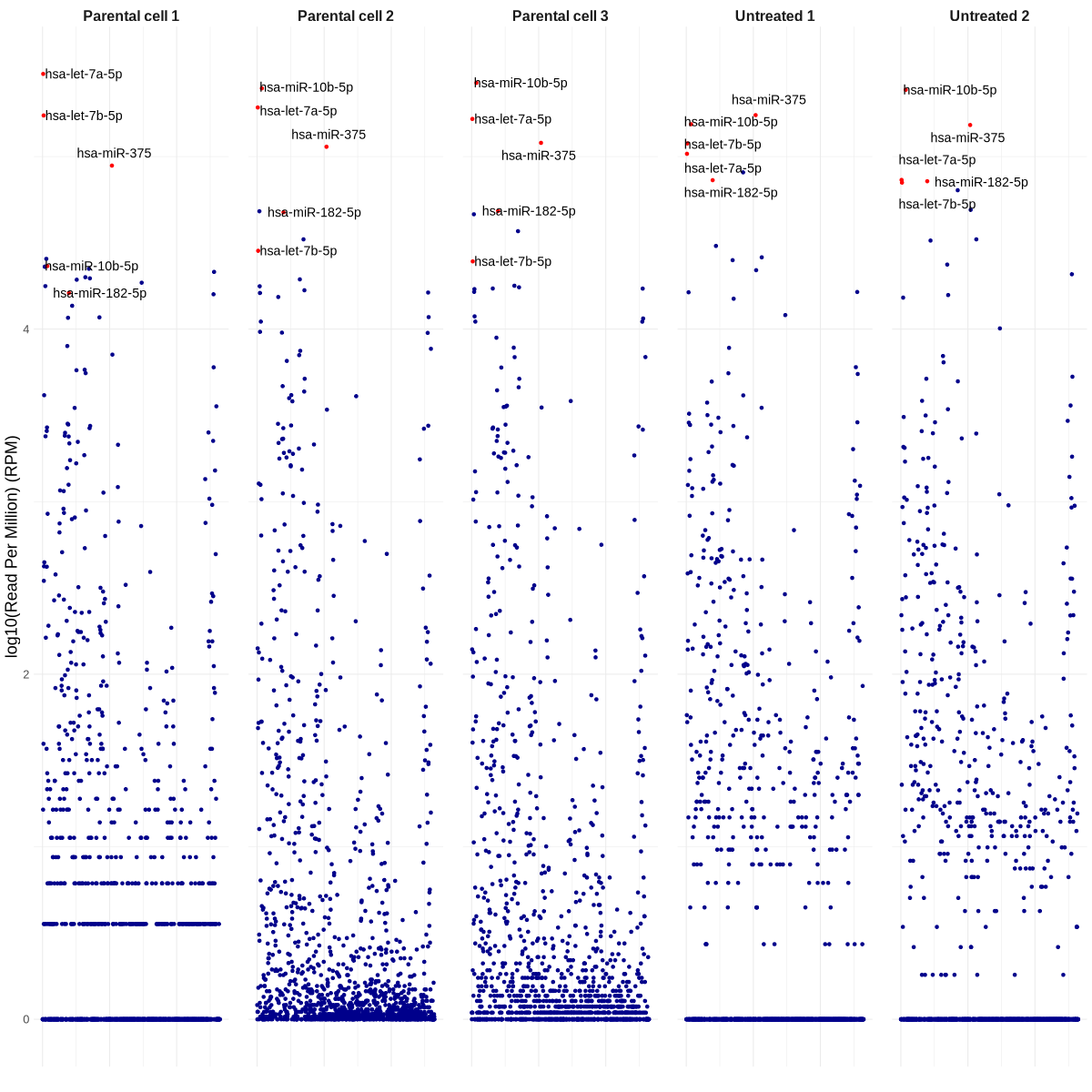
# Step 5: Identify top miRNAs based on mean RPM
top_mirnas <- df %>%
group_by(miRNA) %>%
summarise(mean_RPM = mean(RPM)) %>%
arrange(desc(mean_RPM)) %>%
head(5) %>%
pull(miRNA) # Get the names of top 5 miRNAs
# Step 6: Assign color based on whether the miRNA is top or not
df$color <- ifelse(df$miRNA %in% top_mirnas, "red", "darkblue")
# Rename the sample labels for display
sample_labels <- c(
"parental_cells_1" = "Parental cell 1",
"parental_cells_2" = "Parental cell 2",
"parental_cells_3" = "Parental cell 3",
"untreated_1" = "Untreated 1",
"untreated_2" = "Untreated 2"
)
# Step 7: Plot
png("manhattan_plot_top_miRNAs_based_on_mean_RPM.png", width = 1200, height = 1200)
ggplot(df, aes(x = Position, y = logRPM, color = color)) +
scale_color_manual(values = c("red" = "red", "darkblue" = "darkblue")) +
geom_jitter(width = 0.4) +
geom_text_repel(
data = df %>% filter(miRNA %in% top_mirnas),
aes(label = miRNA),
box.padding = 0.5,
point.padding = 0.5,
segment.color = 'gray50',
size = 5,
max.overlaps = 8,
color = "black"
) +
labs(x = "", y = "log10(Read Per Million) (RPM)") +
facet_wrap(~sample, scales = "free_x", ncol = 5,
labeller = labeller(sample = sample_labels)) +
theme_minimal() +
theme(
axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
legend.position = "none",
text = element_text(size = 16),
axis.title = element_text(size = 18),
strip.text = element_text(size = 16, face = "bold"),
panel.spacing = unit(1.5, "lines") # <-- More space between plots
)
dev.off()
top_mirnas = c("hsa-miR-20a-5p","hsa-miR-93-5p","hsa-let-7g-5p","hsa-miR-30a-5p","hsa-miR-423-5p","hsa-let-7i-5p")
#,"hsa-miR-17-5p","hsa-miR-107","hsa-miR-483-5p","hsa-miR-9-5p","hsa-miR-103a-3p","hsa-miR-30e-5p","hsa-miR-21-5p","hsa-miR-30d-5p")
# Step 6: Assign color based on whether the miRNA is top or not
df$color <- ifelse(df$miRNA %in% top_mirnas, "red", "darkblue")
# Rename the sample labels for display
sample_labels <- c(
"parental_cells_1" = "Parental cell 1",
"parental_cells_2" = "Parental cell 2",
"parental_cells_3" = "Parental cell 3",
"untreated_1" = "Untreated 1",
"untreated_2" = "Untreated 2"
)
# Step 7: Plot
png("manhattan_plot_most_differentially_expressed_miRNAs.png", width = 1200, height = 1200)
ggplot(df, aes(x = Position, y = logRPM, color = color)) +
scale_color_manual(values = c("red" = "red", "darkblue" = "darkblue")) +
geom_jitter(width = 0.4) +
geom_text_repel(
data = df %>% filter(miRNA %in% top_mirnas),
aes(label = miRNA),
box.padding = 0.5,
point.padding = 0.5,
segment.color = 'gray50',
size = 5,
max.overlaps = 8,
color = "black"
) +
labs(x = "", y = "log10(Read Per Million) (RPM)") +
facet_wrap(~sample, scales = "free_x", ncol = 5,
labeller = labeller(sample = sample_labels)) +
theme_minimal() +
theme(
axis.text.x = element_blank(),
axis.ticks.x = element_blank(),
legend.position = "none",
text = element_text(size = 16),
axis.title = element_text(size = 18),
strip.text = element_text(size = 16, face = "bold"),
panel.spacing = unit(1.5, "lines") # <-- More space between plots
)
dev.off()