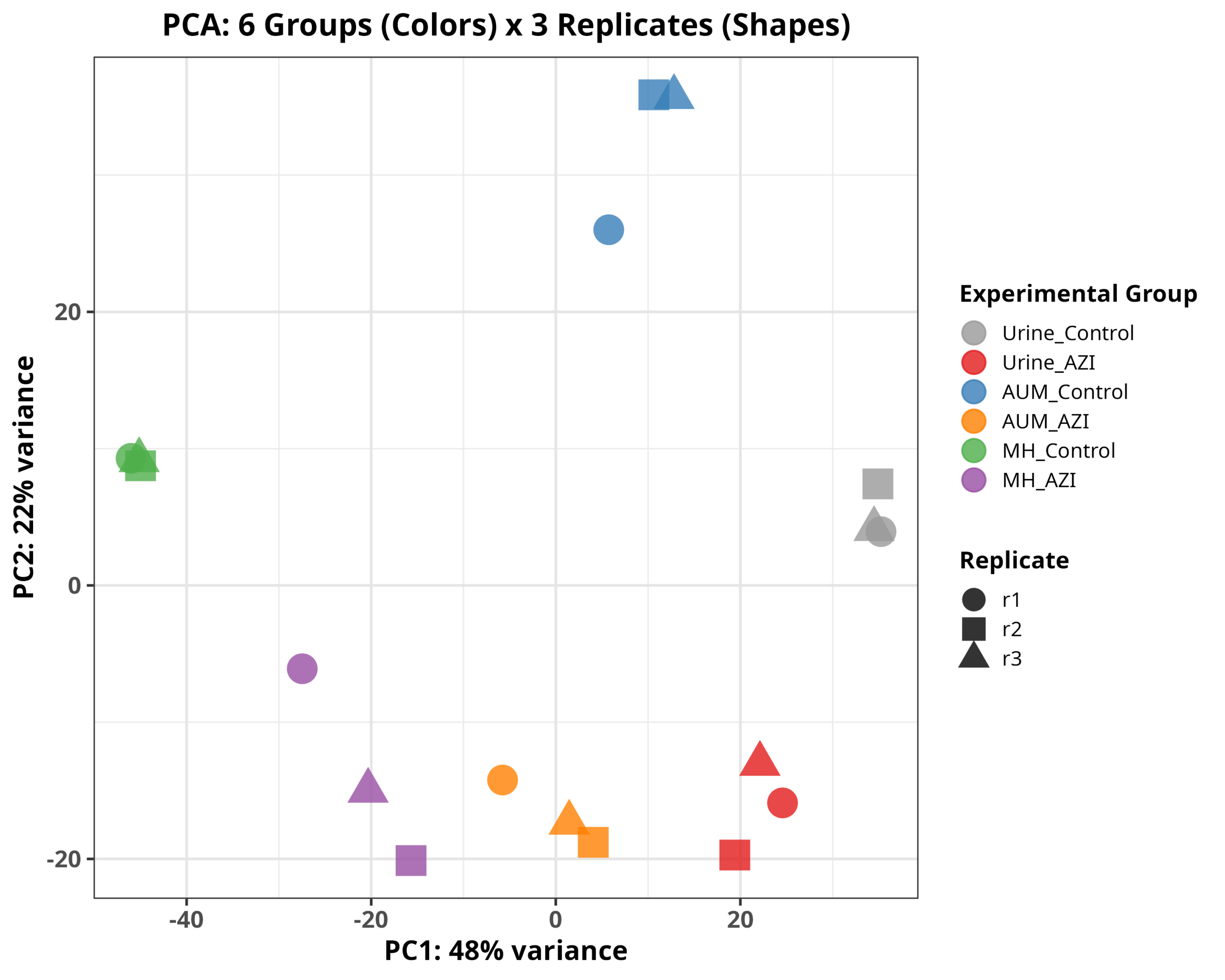
- Results
实验设计框架:
- 环境因素:尿液 (Urine) < AUM 培养基 < MH 培养基(营养梯度递增)
- 处理因素:对照 (Control) vs 阿奇霉素 (AZI)
- 比较组合:共9组比较(3个环境 × AZI效应 + 6组环境间差异)
为便于快速把握整体趋势,我借助AI对9组比较的GO富集结果进行了系统性梳理。该总结提炼了环境梯度与阿奇霉素处理的核心响应模式,我认为对后续机制讨论和论文撰写很有参考价值,具体如下:
📊 分组详细解读
| 🔹 阿奇霉素处理效应(同一环境内) | 比较组 | 上调通路 | 下调通路 | 生物学解读 |
|---|---|---|---|---|
| 01 Urine_AZI vs Control | DNA integration |
无 | 尿液中响应微弱;”DNA 整合”可能提示应激诱导的前噬菌体激活或水平基因转移 | |
| 02 AUM_AZI vs Control | efflux transmembrane transporter, transmembrane transport |
cellular oxidant detoxification |
经典耐药响应:上调外排泵排出阿奇霉素;下调解毒系统以重分配资源 | |
| 03 MH_AZI vs Control | 2Fe-2S cluster binding, carboxylic acid transport |
unfolded protein binding |
代谢适应为主;丰富培养基中蛋白稳态维持良好,无需上调分子伴侣 |
✅ 结论: 阿奇霉素响应具有环境依赖性——仅在营养适中的 AUM 中激活外排泵耐药机制。
| 🔹 基础环境差异(无药物) | 比较组 | 上调通路 | 下调通路 | 生物学解读 |
|---|---|---|---|---|
| 04 AUM_Ctrl vs Urine_Ctrl | enoyl-CoA hydratase activity |
transmembrane transporter, FAD binding |
AUM 支持脂肪酸代谢;尿液需广谱转运蛋白获取稀缺营养 | |
| 05 MH_Ctrl vs Urine_Ctrl | 核糖体/翻译、TCA cycle、NADH dehydrogenase |
siderophore transport、carboxylic acid transport |
MH=生长许可状态:高翻译+呼吸活性;尿液=应激/搜寻状态:铁获取+多样转运 | |
| 06 MH_Ctrl vs AUM_Ctrl | 同#05 的核糖体/能量术语 | siderophore transport、enoyl-CoA hydratase |
确认营养梯度:MH > AUM > Urine;搜寻系统随营养增加而下调 |
| 🔹 阿奇霉素压力下的环境差异 | 比较组 | 上调通路 | 下调通路 | 生物学解读 |
|---|---|---|---|---|
| 07-08: AUM/MH_AZI vs Urine_AZI | 无 | siderophore transport 系列术语 |
即使在药物压力下,尿液环境仍强制细菌维持铁获取系统 | |
| 09 MH_AZI vs AUM_AZI | 核糖体/翻译、GTP binding |
FAD binding、fatty acid beta-oxidation |
药物存在时,MH 仍支持更高翻译活性;AUM+AZI 可能转向脂肪酸分解供能 |
💡 整体观察与后续建议
- 细菌对阿奇霉素的转录响应高度依赖基础营养状态,AUM 环境最易触发经典外排泵耐药,而尿液环境则倾向于进入低代谢/持留状态。
- 铁获取系统(siderophore)与跨膜转运在限制性环境中始终处于核心地位,建议可作为后续机制讨论或靶点验证的重点方向。
- 目前富集分析基于 ORA (
enricher),部分小基因列表的p.adjust可能偏高。若后续需要更高灵敏度,我可随时切换为基于全基因排序的GSEA流程。
-
Preparing raw data for the batch 2 dataset
They are wildtype strains grown in different medium. Urine - human urine AUM - artificial urine medium MHB - Mueller-Hinton broth Urine(人类尿液):pH值、比重、温度、污染物、化学成分、微生物负荷。 AUM(人工尿液培养基):pH值、营养成分、无菌性、渗透压、温度、污染物。 MHB(Mueller-Hinton培养基):pH值、无菌性、营养成分、温度、渗透压、抗生素浓度。 阿奇霉素(Azithromycin,常缩写 AZI)是大环内酯类抗生素的一种。 * 作用机制:主要通过结合细菌核糖体 50S 亚基(23S rRNA),阻止蛋白质合成中的“肽链延伸”,从而抑制细菌生长(多为抑菌作用,某些情况下也可杀菌)。 * 常见适应证:上呼吸道/下呼吸道感染、支原体/衣原体感染、部分皮肤软组织感染等(具体要看地区指南和耐药情况)。 * 特点:半衰期较长、组织分布好,所以常见给药方案是“三日疗法/五日疗法”。但也因为广泛使用,耐药问题比较突出。 * 耐药机制(概念性):常见包括 1. 23S rRNA 甲基化(erm 基因)导致结合位点改变; 2. 外排泵增加(efflux); 3. 核糖体蛋白突变等。 * 注意事项(概念性):可能引起胃肠道不适;少数人有心电图 QT 间期延长风险;和某些药物相互作用需要注意(具体用药应遵医嘱)。 mkdir raw_data; cd raw_data # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-1/AUM-1_1.fq.gz AUM_r4_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-1/AUM-1_2.fq.gz AUM_r4_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-2/AUM-2_1.fq.gz AUM_r5_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-2/AUM-2_2.fq.gz AUM_r5_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-3/AUM-3_1.fq.gz AUM_r6_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-3/AUM-3_2.fq.gz AUM_r6_R2.fq.gz # # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-1/MHB-1_1.fq.gz MH_r4_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-1/MHB-1_2.fq.gz MH_r4_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-2/MHB-2_1.fq.gz MH_r5_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-2/MHB-2_2.fq.gz MH_r5_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-3/MHB-3_1.fq.gz MH_r6_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-3/MHB-3_2.fq.gz MH_r6_R2.fq.gz # # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-1/Urine-1_1.fq.gz Urine_r4_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-1/Urine-1_2.fq.gz Urine_r4_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-2/Urine-2_1.fq.gz Urine_r5_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-2/Urine-2_2.fq.gz Urine_r5_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-3/Urine-3_1.fq.gz Urine_r6_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-3/Urine-3_2.fq.gz Urine_r6_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-1/AUM-1_1.fq.gz AUM_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-1/AUM-1_2.fq.gz AUM_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-2/AUM-2_1.fq.gz AUM_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-2/AUM-2_2.fq.gz AUM_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-3/AUM-3_1.fq.gz AUM_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-3/AUM-3_2.fq.gz AUM_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-1/AUM-AZI-1_1.fq.gz AUM-AZI_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-1/AUM-AZI-1_2.fq.gz AUM-AZI_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-2/AUM-AZI-2_1.fq.gz AUM-AZI_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-2/AUM-AZI-2_2.fq.gz AUM-AZI_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-3/AUM-AZI-3_1.fq.gz AUM-AZI_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-3/AUM-AZI-3_2.fq.gz AUM-AZI_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-1/MH-1_1.fq.gz MH_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-1/MH-1_2.fq.gz MH_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-2/MH-2_1.fq.gz MH_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-2/MH-2_2.fq.gz MH_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-3/MH-3_1.fq.gz MH_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-3/MH-3_2.fq.gz MH_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-1/MH-AZI-1_1.fq.gz MH-AZI_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-1/MH-AZI-1_2.fq.gz MH-AZI_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-2/MH-AZI-2_1.fq.gz MH-AZI_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-2/MH-AZI-2_2.fq.gz MH-AZI_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-3/MH-AZI-3_1.fq.gz MH-AZI_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-3/MH-AZI-3_2.fq.gz MH-AZI_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-1/Urine-1_1.fq.gz Urine_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-1/Urine-1_2.fq.gz Urine_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-2/Urine-2_1.fq.gz Urine_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-2/Urine-2_2.fq.gz Urine_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-3/Urine-3_1.fq.gz Urine_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-3/Urine-3_2.fq.gz Urine_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-1/Urine-AZI-1_1.fq.gz Urine-AZI_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-1/Urine-AZI-1_2.fq.gz Urine-AZI_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-2/Urine-AZI-2_1.fq.gz Urine-AZI_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-2/Urine-AZI-2_2.fq.gz Urine-AZI_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-3/Urine-AZI-3_1.fq.gz Urine-AZI_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-3/Urine-AZI-3_2.fq.gz Urine-AZI_r3_R2.fq.gz -
Preparing the directory trimmed
mkdir trimmed trimmed_unpaired; for sample_id in AUM_r1 AUM_r2 AUM_r3 AUM_r4 AUM_r5 AUM_r6 Urine_r1 Urine_r2 Urine_r3 Urine_r4 Urine_r5 Urine_r6 MH_r1 MH_r2 MH_r3 MH_r4 MH_r5 MH_r6 AUM-AZI_r1 AUM-AZI_r2 AUM-AZI_r3 Urine-AZI_r1 Urine-AZI_r2 Urine-AZI_r3 MH-AZI_r1 MH-AZI_r2 MH-AZI_r3; do \ java -jar /home/jhuang/Tools/Trimmomatic-0.36/trimmomatic-0.36.jar PE -threads 100 raw_data/${sample_id}_R1.fq.gz raw_data/${sample_id}_R2.fq.gz trimmed/${sample_id}_R1.fq.gz trimmed_unpaired/${sample_id}_R1.fq.gz trimmed/${sample_id}_R2.fq.gz trimmed_unpaired/${sample_id}_R2.fq.gz ILLUMINACLIP:/home/jhuang/Tools/Trimmomatic-0.36/adapters/TruSeq3-PE-2.fa:2:30:10:8:TRUE LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 AVGQUAL:20; done 2> trimmomatic_pe.log; done -
Preparing samplesheet.csv
sample,fastq_1,fastq_2,strandedness Urine_r1,Urine_r1_R1.fq.gz,Urine_r1_R2.fq.gz,auto Urine_r2,Urine_r2_R1.fq.gz,Urine_r2_R2.fq.gz,auto Urine_r3,Urine_r3_R1.fq.gz,Urine_r3_R2.fq.gz,auto Urine_r4,Urine_r4_R1.fq.gz,Urine_r4_R2.fq.gz,auto Urine_r5,Urine_r5_R1.fq.gz,Urine_r5_R2.fq.gz,auto Urine_r6,Urine_r6_R1.fq.gz,Urine_r6_R2.fq.gz,auto AUM_r1,AUM_r1_R1.fq.gz,AUM_r1_R2.fq.gz,auto AUM_r2,AUM_r2_R1.fq.gz,AUM_r2_R2.fq.gz,auto AUM_r3,AUM_r3_R1.fq.gz,AUM_r3_R2.fq.gz,auto AUM_r4,AUM_r4_R1.fq.gz,AUM_r4_R2.fq.gz,auto AUM_r5,AUM_r5_R1.fq.gz,AUM_r5_R2.fq.gz,auto AUM_r6,AUM_r6_R1.fq.gz,AUM_r6_R2.fq.gz,auto MH_r1,MH_r1_R1.fq.gz,MH_r1_R2.fq.gz,auto MH_r2,MH_r2_R1.fq.gz,MH_r2_R2.fq.gz,auto MH_r3,MH_r3_R1.fq.gz,MH_r3_R2.fq.gz,auto MH_r4,MH_r4_R1.fq.gz,MH_r4_R2.fq.gz,auto MH_r5,MH_r5_R1.fq.gz,MH_r5_R2.fq.gz,auto MH_r6,MH_r6_R1.fq.gz,MH_r6_R2.fq.gz,auto Urine-AZI_r1,Urine-AZI_r1_R1.fq.gz,Urine-AZI_r1_R2.fq.gz,auto Urine-AZI_r2,Urine-AZI_r2_R1.fq.gz,Urine-AZI_r2_R2.fq.gz,auto Urine-AZI_r3,Urine-AZI_r3_R1.fq.gz,Urine-AZI_r3_R2.fq.gz,auto AUM-AZI_r1,AUM-AZI_r1_R1.fq.gz,AUM-AZI_r1_R2.fq.gz,auto AUM-AZI_r2,AUM-AZI_r2_R1.fq.gz,AUM-AZI_r2_R2.fq.gz,auto AUM-AZI_r3,AUM-AZI_r3_R1.fq.gz,AUM-AZI_r3_R2.fq.gz,auto MH-AZI_r1,MH-AZI_r1_R1.fq.gz,MH-AZI_r1_R2.fq.gz,auto MH-AZI_r2,MH-AZI_r2_R1.fq.gz,MH-AZI_r2_R2.fq.gz,auto MH-AZI_r3,MH-AZI_r3_R1.fq.gz,MH-AZI_r3_R2.fq.gz,auto -
Preparing CP059040.fasta, CP059040_gene.gff3 and CP059040.bed and nextflow run
# -- DEBUG_1 (CDS --> exon in CP059040.gff) -- #Checking the record (see below) in results/genome/CP059040.gtf #In ./results/genome/CP059040.gtf e.g. "CP059040.1 Genbank transcript 1 1398 . + . transcript_id "gene-H0N29_00005"; gene_id "gene-H0N29_00005"; gene_name "dnaA"; Name "dnaA"; gbkey "Gene"; gene "dnaA"; gene_biotype "protein_coding"; locus_tag "H0N29_00005";" #--featurecounts_feature_type 'transcript' returns only the tRNA results #Since the tRNA records have "transcript and exon". In gene records, we have "transcript and CDS". replace the CDS with exon grep -P "\texon\t" CP059040.gff | sort | wc -l #96 grep -P "cmsearch\texon\t" CP059040.gff | wc -l #=10 ignal recognition particle sRNA small typ, transfer-messenger RNA, 5S ribosomal RNA grep -P "Genbank\texon\t" CP059040.gff | wc -l #=12 16S and 23S ribosomal RNA grep -P "tRNAscan-SE\texon\t" CP059040.gff | wc -l #tRNA 74 wc -l star_salmon/AUM_r3/quant.genes.sf #--featurecounts_feature_type 'transcript' results in 96 records! grep -P "\tCDS\t" CP059040.gff | wc -l #3701 sed 's/\tCDS\t/\texon\t/g' CP059040.gff > CP059040_m.gff grep -P "\texon\t" CP059040_m.gff | sort | wc -l #3797 # -- DEBUG_2: combination of 'CP059040_m.gff' and 'exon' results in ERROR, using 'transcript' instead! --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024/CP059040_m.gff" --featurecounts_feature_type 'transcript' # ---- SUCCESSFUL with directly downloaded gff3 and fasta from NCBI using docker after replacing 'CDS' with 'exon' ---- (host_env) mv trimmed/*.fastq.gz . (host_env) nextflow run nf-core/rnaseq -r 3.14.0 -profile docker \ --input samplesheet.csv --outdir results --fasta "/home/jhuang/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/CP059040.fasta" --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/CP059040_m.gff" -resume --max_cpus 90 --max_memory 900.GB --max_time 2400.h --save_align_intermeds --save_unaligned --save_reference --aligner ‘star_salmon’ --gtf_group_features ‘gene_id’ --gtf_extra_attributes ‘gene_name’ --featurecounts_group_type ‘gene_biotype’ --featurecounts_feature_type ‘transcript’ # -- DEBUG_3: make sure the header of fasta is the same to the *_m.gff file -
Import data and pca-plot
# ============================================================================== # ADAPTED PIPELINE: 6 Groups (Urine/AUM/MH ± AZI) -> Counts Export -> PCA # ============================================================================== # 1️⃣ LOAD LIBRARIES ------------------------------------------------------------ suppressPackageStartupMessages({ library(DESeq2) library(tximport) library(dplyr) library(ggplot2) library(ggrepel) library(edgeR) # For robust CPM calculation library(openxlsx) # For Excel export }) # 2️⃣ SET WORKING DIRECTORY & DEFINE SAMPLES ------------------------------------ setwd("/mnt/md1/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_on_ATCC19606/results/star_salmon") files <- c( "AUM-AZI_r1" = "./AUM-AZI_r1/quant.sf", "AUM-AZI_r2" = "./AUM-AZI_r2/quant.sf", "AUM-AZI_r3" = "./AUM-AZI_r3/quant.sf", "AUM_r1" = "./AUM_r1/quant.sf", "AUM_r2" = "./AUM_r2/quant.sf", "AUM_r3" = "./AUM_r3/quant.sf", "MH-AZI_r1" = "./MH-AZI_r1/quant.sf", "MH-AZI_r2" = "./MH-AZI_r2/quant.sf", "MH-AZI_r3" = "./MH-AZI_r3/quant.sf", "MH_r1" = "./MH_r1/quant.sf", "MH_r2" = "./MH_r2/quant.sf", "MH_r3" = "./MH_r3/quant.sf", "Urine-AZI_r1" = "./Urine-AZI_r1/quant.sf", "Urine-AZI_r2" = "./Urine-AZI_r2/quant.sf", "Urine-AZI_r3" = "./Urine-AZI_r3/quant.sf", "Urine_r1" = "./Urine_r1/quant.sf", "Urine_r2" = "./Urine_r2/quant.sf", "Urine_r3" = "./Urine_r3/quant.sf" ) # 3️⃣ AUTOMATED METADATA PARSING ----------------------------------------------- # Dynamically extracts Media, Treatment, and Combined Group from filenames samples <- names(files) colData <- data.frame( media = factor(gsub("-.*", "", samples)), treatment = factor(ifelse(grepl("AZI", samples), "AZI", "Control")), group = factor(paste(gsub("-.*", "", samples), ifelse(grepl("AZI", samples), "AZI", "Control"), sep = "_")), replicate = as.numeric(gsub(".*r", "", samples)), row.names = samples, stringsAsFactors = FALSE ) # 4️⃣ IMPORT & SUMMARIZE TO GENE LEVEL ----------------------------------------- tx2gene <- read.table("salmon_tx2gene.tsv", header = FALSE, stringsAsFactors = FALSE) colnames(tx2gene) <- c("transcript_id", "gene_id", "gene_name") tx2gene_geneonly <- tx2gene[, c("transcript_id", "gene_id")] # Direct gene-level import (faster & standard for DESeq2) txi <- tximport(files, type = "salmon", tx2gene = tx2gene_geneonly, txOut = FALSE) # Build DESeq2 object dds <- DESeqDataSetFromTximport(txi, colData = colData, design = ~ group) # Optional: Pre-filter low-count genes (improves VST & PCA stability) keep <- rowSums(counts(dds) >= 10) >= 3 dds <- dds[keep, ] # 5️⃣ EXPORT RAW COUNTS & CPM ------------------------------------------------- counts_data <- as.data.frame(counts(dds, normalized = FALSE)) counts_data$gene_id <- rownames(counts_data) # Merge gene names tx2gene_unique <- unique(tx2gene[, c("gene_id", "gene_name")]) counts_data <- merge(counts_data, tx2gene_unique, by = "gene_id", all.x = TRUE) count_cols <- setdiff(colnames(counts_data), c("gene_id", "gene_name")) counts_data <- counts_data[, c("gene_id", "gene_name", count_cols)] # Calculate CPM (edgeR handles library size normalization automatically) cpm_matrix <- edgeR::cpm(as.matrix(counts_data[, count_cols])) cpm_counts <- cbind(counts_data[, c("gene_id", "gene_name")], as.data.frame(cpm_matrix)) # Save tables write.csv(counts_data, "gene_raw_counts.csv", row.names = FALSE) write.xlsx(counts_data, "gene_raw_counts.xlsx", row.names = FALSE) write.xlsx(cpm_counts, "gene_cpm_counts.xlsx", row.names = FALSE) cat("✅ Count tables exported successfully.\n") # ============================================================================== # 6️⃣ PCA PLOTTING ------------------------------------------------------------- # ============================================================================== vsd <- vst(dds, blind = FALSE) pca_data <- plotPCA(vsd, intgroup = c("media", "treatment", "group"), returnData = TRUE) percent_var <- round(100 * attr(pca_data, "percentVar")) base_theme <- theme_bw(base_size = 12) + theme(plot.title = element_text(hjust = 0.5, face = "bold", size = 13), legend.position = "right", legend.title = element_text(face = "bold"), panel.grid.major = element_line(color = "grey90"), panel.grid.minor = element_blank()) # --- Plot 1: By Culture Media --- p1 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = media)) + geom_point(size = 3, alpha = 0.8) + geom_text_repel(aes(label = name), size = 2.5, max.overlaps = 20, show.legend = FALSE) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: Samples Colored by Media", color = "Media") + base_theme ggsave("01_PCA_by_Media.png", p1, width = 8, height = 6, dpi = 300) # --- Plot 2: By Treatment (AZI vs Control) --- p2 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = treatment)) + geom_point(size = 3, alpha = 0.8) + geom_text_repel(aes(label = name), size = 2.5, max.overlaps = 20, show.legend = FALSE) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: Samples Colored by Azithromycin Treatment", color = "Treatment") + base_theme ggsave("02_PCA_by_Treatment.png", p2, width = 8, height = 6, dpi = 300) # --- Plot 3: Combined Groups (Labeled) --- p3 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = group)) + geom_point(size = 3, alpha = 0.8) + geom_text_repel(aes(label = name), size = 2.2, max.overlaps = 30, box.padding = 0.3) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: Combined Media × Treatment Groups", color = "Group") + base_theme + theme(legend.position = "bottom") ggsave("03_PCA_CombinedGroups.png", p3, width = 9, height = 7, dpi = 300) # --- Plot 4: 95% Confidence Ellipses (by Media) --- p4 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = media, fill = media)) + geom_point(size = 3, alpha = 0.7) + stat_ellipse(level = 0.95, alpha = 0.2, geom = "polygon", show.legend = FALSE) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: 95% Confidence Ellipses by Media", color = "Media", fill = "Media") + base_theme ggsave("04_PCA_Ellipses.png", p4, width = 8, height = 6, dpi = 300) message("✅ All 4 PCA plots saved to working directory!") # 1. Generate PCA Data vsd <- vst(dds, blind = FALSE) pca_data <- plotPCA(vsd, intgroup = c("media", "treatment", "replicate"), returnData = TRUE) # 2. FIX: Clean the 'media' column (remove _r1, _r2, _r3 suffix) pca_data$media_clean <- gsub("_r[1-3]$", "", pca_data$media) # 3. Create Group Variable with cleaned media names pca_data$Group <- factor(paste(pca_data$media_clean, pca_data$treatment, sep = "_"), levels = c("Urine_Control", "Urine_AZI", "AUM_Control", "AUM_AZI", "MH_Control", "MH_AZI")) # 4. Convert replicate to factor for shape mapping pca_data$replicate <- factor(pca_data$replicate, levels = c(1, 2, 3), labels = c("r1", "r2", "r3")) # 5. Define 6 Colors my_colors <- c( "Urine_Control" = "#999999", "Urine_AZI" = "#E41A1C", "AUM_Control" = "#377EB8", "AUM_AZI" = "#FF7F00", "MH_Control" = "#4DAF4A", "MH_AZI" = "#984EA3" ) percent_var <- round(100 * attr(pca_data, "percentVar")) # 6. Plotting p <- ggplot(pca_data, aes(x = PC1, y = PC2, color = Group, shape = replicate)) + geom_point(size = 8, alpha = 0.8) + scale_color_manual(values = my_colors) + scale_shape_manual(values = c("r1" = 16, "r2" = 15, "r3" = 17)) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: 6 Groups (Colors) x 3 Replicates (Shapes)", color = "Experimental Group", shape = "Replicate") + theme_bw(base_size = 16) + theme( axis.text = element_text(face = "bold", size = 14), axis.title = element_text(face = "bold", size = 16), legend.title = element_text(face = "bold", size = 14), legend.text = element_text(size = 12), plot.title = element_text(hjust = 0.5, face = "bold", size = 18), panel.grid.major = element_line(color = "grey90") ) + guides(color = guide_legend(override.aes = list(size = 6)), shape = guide_legend(override.aes = list(size = 6))) ggsave("PCA_Group_x_Replicate.png", p, width = 10, height = 8, dpi = 300) # Verify the fix print(table(pca_data$Group)) # 1. PCA Data Extraction vsd <- vst(dds, blind = FALSE) pca_data <- plotPCA(vsd, intgroup = c("media", "treatment", "replicate"), returnData = TRUE) # 2. CRITICAL FIX: Clean 'media' column to remove replicate suffixes (_r1, _r2, _r3) pca_data$media_clean <- gsub("_r[1-3]$", "", pca_data$media) # 3. Create Group & Replicate identifiers pca_data$Group <- paste(pca_data$media_clean, pca_data$treatment, sep = "_") pca_data$Replicate <- sub(".*_(r\\d+)$", "\\1", pca_data$name, ignore.case = TRUE) # Define logical ordering for consistent legend layout group_order <- c("Urine_Control", "Urine_AZI", "AUM_Control", "AUM_AZI", "MH_Control", "MH_AZI") pca_data$Group <- factor(pca_data$Group, levels = group_order) pca_data$Replicate <- factor(pca_data$Replicate, levels = c("r1", "r2", "r3")) # Generate SampleID with explicit ordering (Group1:r1,r2,r3 -> Group2:r1,r2,r3 ...) pca_data$SampleID <- factor(paste(pca_data$Group, pca_data$Replicate, sep = "_"), levels = paste(rep(group_order, each = 3), rep(c("r1", "r2", "r3"), times = 6), sep = "_")) # 4. Define 18 Colors (6 groups × 3 progressive shades) sample_colors <- c( "Urine_Control_r1" = "#1B5E77", "Urine_Control_r2" = "#1B9E77", "Urine_Control_r3" = "#66CCB5", "Urine_AZI_r1" = "#B34A00", "Urine_AZI_r2" = "#D95F02", "Urine_AZI_r3" = "#F2A65A", "AUM_Control_r1" = "#4A3D7A", "AUM_Control_r2" = "#7570B3", "AUM_Control_r3" = "#B3B0D9", "AUM_AZI_r1" = "#B31A6A", "AUM_AZI_r2" = "#E7298A", "AUM_AZI_r3" = "#F285B8", "MH_Control_r1" = "#4A7A15", "MH_Control_r2" = "#66A61E", "MH_Control_r3" = "#A3D66B", "MH_AZI_r1" = "#7A5A15", "MH_AZI_r2" = "#A6761D", "MH_AZI_r3" = "#D6B86B" ) percent_var <- round(100 * attr(pca_data, "percentVar")) # 5. Plotting p <- ggplot(pca_data, aes(x = PC1, y = PC2, color = SampleID)) + geom_point(size = 5, shape = 16) + scale_color_manual(values = sample_colors) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: 18 Samples (Grouped by Color Shades)", color = "Sample ID") + theme_bw() + theme( plot.background = element_rect(fill = "white", color = NA), panel.background = element_rect(fill = "white", color = "grey85"), panel.grid.major = element_line(color = "grey90"), panel.grid.minor = element_blank(), legend.position = "right", legend.title = element_text(face = "bold", size = 11), legend.text = element_text(size = 9), axis.text = element_text(color = "black", size = 10), axis.title = element_text(face = "bold", size = 11), plot.margin = margin(0.5, 0.5, 0.5, 0.5, "cm") # Prevents legend cutoff ) + guides(color = guide_legend(override.aes = list(size = 6), nrow = 6, title.position = "top")) # 6. Quick validation before saving cat("Sample mapping check:\n") print(table(pca_data$SampleID)) # 7. Save & Display ggsave("PCA_18_Samples_GroupedColors.png", p, width = 11, height = 8, dpi = 300) message("✅ All 2 PCA plots saved to working directory!") -
Perform DEG analysis
# In DESeq2, an NA in the pvalue column (and typically also in padj) is intentional and indicates that the gene was excluded from statistical testing. # Key Changes Made: # 1. NA Handling: Added pvalue = ifelse(is.na(pvalue), 1, pvalue) and padj = ifelse(is.na(padj), 1, padj) in the mutate block. This converts statistical NAs (usually from low counts or outliers) to 1, marking them as "Not Significant". # 2. Ordering: Genes with NA (now 1) will sort to the bottom of your Excel sheets and appear at y=0 on volcano plots, which is the correct visual representation for non-significant genes. # 3. Plot Safety: The padj_plot column still handles padj == 0 by converting it to 1e-305 to prevent -log10(0) = Inf errors in the volcano plot.
Rscript perform_DEG_analysis.R-
TRY summarizing the process of 8.2 into a R-script, but does not work! Then using the code and process described in 8 for KEGG and GO enrichments
Rscript batch_enrichment.R 🔑 Key Improvements Made: * Exclusive Blast2GO for GO/EC: go_terms and ec_terms are parsed directly from blast2go_annot.annot2_. EggNOG is never used for GO/EC, eliminating .x/.y suffix conflicts entirely. * Selective EggNOG Join: eggnog_kegg only contains GeneID and KEGG_ko. This prevents bringing in EggNOG's sparse GO/EC columns into your main table. * Fixed Syntax & Piping: Corrected all % >% → %>%, fixed missing assignments, cleaned up tryCatch blocks, and removed trailing spaces in file paths that would cause file.exists() failures. * Streamlined go_annot_tbl: Now pulls directly from the Blast2GO-derived GOs column in res_annot, ensuring clusterProfiler::enricher() receives clean, non-colliding data. * Robust NA Handling: Explicitly replaces NA with "-" for KEGG_ko, GOs, and EC after joins, so downstream filtering (filter(GOs != "-")) works reliably. * "enrichKEGG(gene = kos, organism = 'ko', pvalueCutoff = KEGG_P_CUT)" This function internally uses the KEGG REST API (https://rest.kegg.jp/) to download pathway-gene mappings. The actual HTTP requests are handled by the KEGGREST package (a dependency of clusterProfiler). -
KEGG and GO annotations in non-model organisms
8.1. Assign KEGG and GO Terms (see diagram above)
Since your organism is non-model, standard R databases (org.Hs.eg.db, etc.) won’t work. You’ll need to manually retrieve KEGG and GO annotations.
* Preparing file 1 eggnog_out.emapper.annotations.txt for the R-code below: (KEGG Terms): EggNog based on orthology and phylogenies
EggNOG-mapper assigns both KEGG Orthology (KO) IDs and GO terms.
Install EggNOG-mapper:
mamba create -n eggnog_env python=3.8 eggnog-mapper -c conda-forge -c bioconda #eggnog-mapper_2.1.12
mamba activate eggnog_env
Run annotation:
#diamond makedb --in eggnog6.prots.faa -d eggnog_proteins.dmnd
mkdir /home/jhuang/mambaforge/envs/eggnog_env/lib/python3.8/site-packages/data/
download_eggnog_data.py --dbname eggnog.db -y --data_dir /home/jhuang/mambaforge/envs/eggnog_env/lib/python3.8/site-packages/data/
#NOT_WORKING: emapper.py -i CP059040_gene.fasta -o eggnog_dmnd_out --cpu 60 -m diamond[hmmer,mmseqs] --dmnd_db /home/jhuang/REFs/eggnog_data/data/eggnog_proteins.dmnd
python ~/Scripts/update_fasta_header.py CP059040_protein_.fasta CP059040_protein.fasta
emapper.py -i CP059040_protein.fasta -o eggnog_out --cpu 60 --resume
#----> result annotations.tsv: Contains KEGG, GO, and other functional annotations.
#----> 470.IX87_14445:
* 470 likely refers to the organism or strain (e.g., Acinetobacter baumannii ATCC 19606 or another related strain).
* IX87_14445 would refer to a specific gene or protein within that genome.
Extract KEGG KO IDs from annotations.emapper.annotations.
* Preparing file 2 blast2go_annot.annot2_ for the R-code below:
- Basic (GO Terms from 'Blast2GO 5 Basic', saved in blast2go_annot.annot): Using Blast/Diamond + Blast2GO_GUI based on sequence alignment + GO mapping
* 'Load protein sequences' (Tags: NONE, generated columns: Nr, SeqName) -->
* Buttons 'blast' (Tags: BLASTED, generated columns: Description, Length, #Hits, e-Value, sim mean),
* Button 'mapping' (Tags: MAPPED, generated columns: #GO, GO IDs, GO Names), "Mapping finished - Please proceed now to annotation."
* Button 'annot' (Tags: ANNOTATED, generated columns: Enzyme Codes, Enzyme Names), "Annotation finished."
* Used parameter 'Annotation CutOff': The Blast2GO Annotation Rule seeks to find the most specific GO annotations with a certain level of reliability. An annotation score is calculated for each candidate GO which is composed by the sequence similarity of the Blast Hit, the evidence code of the source GO and the position of the particular GO in the Gene Ontology hierarchy. This annotation score cutoff select the most specific GO term for a given GO branch which lies above this value.
* Used parameter 'GO Weight' is a value which is added to Annotation Score of a more general/abstract Gene Ontology term for each of its more specific, original source GO terms. In this case, more general GO terms which summarise many original source terms (those ones directly associated to the Blast Hits) will have a higher Annotation Score.
- Advanced (GO Terms from 'Blast2GO 5 Basic'): Interpro based protein families / domains --> Button interpro
* Button 'interpro' (Tags: INTERPRO, generated columns: InterPro IDs, InterPro GO IDs, InterPro GO Names) --> "InterProScan Finished - You can now merge the obtained GO Annotations."
- MERGE the results of InterPro GO IDs (advanced) to GO IDs (basic) and generate final GO IDs, saved in blast2go_annot.annot2
* Button 'interpro'/'Merge InterProScan GOs to Annotation' --> "Merge (add and validate) all GO terms retrieved via InterProScan to the already existing GO annotation." --> "Finished merging GO terms from InterPro with annotations. Maybe you want to run ANNEX (Annotation Augmentation)."
* (NOT_USED) Button 'annot'/'ANNEX' --> "ANNEX finished. Maybe you want to do the next step: Enzyme Code Mapping."
- PREPARING go_terms and ec_terms: annot_* file:
cut -f1-2 -d$'\t' blast2go_annot.annot2 > blast2go_annot.annot2_
8.2. Perform KEGG and GO Enrichment in R
# SAVE the complete sheet from the Excel-files to csv-format.
Rscript batch_enrichment.R (NOT_WORKING, USING the old R code below)!
# Preparing the input csv-format from Excel, e.g.
#Replace with DEG_02_AUM_AZI_vs_Control.csv
#Replace with DEG_02_AUM_AZI_vs_Control.csv
#Replace with DEG_03_MH_AZI_vs_Control.csv
#Replace with DEG_04_AUM_vs_Urine_Control.csv
#Replace with DEG_05_MH_vs_Urine_Control.csv
#Replace with DEG_06_MH_vs_AUM_Control.csv
#Replace with DEG_07_AUM_vs_Urine_AZI.csv
#Replace with DEG_08_MH_vs_Urine_AZI.csv
#Replace with DEG_09_MH_vs_AUM_AZI.csv
#BiocManager::install("GO.db")
#BiocManager::install("AnnotationDbi")
# Load required libraries
library(openxlsx) # For Excel file handling
library(dplyr) # For data manipulation
library(tidyr)
library(stringr)
library(clusterProfiler) # For KEGG and GO enrichment analysis
#library(org.Hs.eg.db) # Replace with appropriate organism database
library(GO.db)
library(AnnotationDbi)
setwd("~/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_on_ATCC19606/results/star_salmon/DEG_Results_6Groups")
# 1. Blast2GO: Extract GO & EC terms (Primary source)
annot_df <- read.table("/home/jhuang/b2gWorkspace_Tam_RNAseq_2024/blast2go_annot.annot2_",
header = FALSE, sep = "\t", stringsAsFactors = FALSE, fill = TRUE)
colnames(annot_df) <- c("GeneID", "Term")
go_terms <- annot_df %>%
filter(grepl("^GO:", Term)) %>%
group_by(GeneID) %>%
summarize(GOs = paste(Term, collapse = ","), .groups = "drop")
ec_terms <- annot_df %>%
filter(grepl("^EC:", Term)) %>%
group_by(GeneID) %>%
summarize(EC = paste(Term, collapse = ","), .groups = "drop")
# Load the results
res <- read.csv("DEG_09_MH_vs_AUM_AZI.csv")
# Replace empty GeneName with modified GeneID
res$GeneName <- ifelse(
res$GeneName == "" | is.na(res$GeneName),
gsub("gene-", "", res$GeneID),
res$GeneName
)
# Remove duplicated genes by selecting the gene with the smallest padj
duplicated_genes <- res[duplicated(res$GeneName), "GeneName"]
res <- res %>%
group_by(GeneName) %>%
slice_min(padj, with_ties = FALSE) %>%
ungroup()
res <- as.data.frame(res)
# Sort res first by padj (ascending) and then by log2FoldChange (descending)
res <- res[order(res$padj, -res$log2FoldChange), ]
# Read eggnog annotations
eggnog_data <- read.delim("~/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_on_ATCC19606/eggnog_out.emapper.annotations.txt", header = TRUE, sep = "\t")
# Remove the "gene-" prefix from GeneID in res to match eggnog 'query' format
res$GeneID <- gsub("gene-", "", res$GeneID)
# Merge eggnog data with res based on GeneID
res <- res %>%
left_join(eggnog_data, by = c("GeneID" = "query"))
# Merge with the res dataframe
# Perform the left joins and rename columns
res_updated <- res %>%
left_join(go_terms, by = "GeneID") %>%
left_join(ec_terms, by = "GeneID") %>% dplyr::select(-EC.x, -GOs.x) %>% dplyr::rename(EC = EC.y, GOs = GOs.y)
# Filter up- and down-regulated genes
up_regulated <- res_updated[res_updated$log2FoldChange > 2 & res_updated$padj < 0.05, ]
down_regulated <- res_updated[res_updated$log2FoldChange < -2 & res_updated$padj < 0.05, ]
# Create a new workbook
wb <- createWorkbook()
addWorksheet(wb, "Complete_Data")
writeData(wb, "Complete_Data", res_updated)
addWorksheet(wb, "Up_Regulated")
writeData(wb, "Up_Regulated", up_regulated)
addWorksheet(wb, "Down_Regulated")
writeData(wb, "Down_Regulated", down_regulated)
saveWorkbook(wb, "Gene_Expression_with_Annotations_09_MH_vs_AUM_AZI.xlsx", overwrite = TRUE)
# Set GeneName as row names after the join
rownames(res_updated) <- res_updated$GeneName
res_updated <- res_updated %>% dplyr::select(-GeneName)
# ---------------------------------------------------------
# ---- Perform KEGG enrichment analysis (up_regulated) ----
gene_list_kegg_up <- up_regulated$KEGG_ko
gene_list_kegg_up <- gsub("ko:", "", gene_list_kegg_up)
kegg_enrichment_up <- enrichKEGG(gene = gene_list_kegg_up, organism = 'ko')
# -- convert the GeneID (Kxxxxxx) to the true GeneID --
# Step 0: Create KEGG to GeneID mapping
kegg_to_geneid_up <- up_regulated %>%
dplyr::select(KEGG_ko, GeneID) %>%
filter(!is.na(KEGG_ko)) %>% # Remove missing KEGG KO entries
mutate(KEGG_ko = str_remove(KEGG_ko, "ko:")) # Remove 'ko:' prefix if present
# Step 1: Clean KEGG_ko values (separate multiple KEGG IDs)
kegg_to_geneid_clean <- kegg_to_geneid_up %>%
mutate(KEGG_ko = str_remove_all(KEGG_ko, "ko:")) %>% # Remove 'ko:' prefixes
separate_rows(KEGG_ko, sep = ",") %>% # Ensure each KEGG ID is on its own row
filter(KEGG_ko != "-") %>% # Remove invalid KEGG IDs ("-")
distinct() # Remove any duplicate mappings
# Step 2.1: Expand geneID column in kegg_enrichment_up
expanded_kegg <- kegg_enrichment_up %>%
as.data.frame() %>%
separate_rows(geneID, sep = "/") %>% # Split multiple KEGG IDs (Kxxxxx)
left_join(kegg_to_geneid_clean, by = c("geneID" = "KEGG_ko"), relationship = "many-to-many") %>% # Explicitly handle many-to-many
distinct() %>% # Remove duplicate matches
group_by(ID) %>%
summarise(across(everything(), ~ paste(unique(na.omit(.)), collapse = "/")), .groups = "drop") # Re-collapse results
#dplyr::glimpse(expanded_kegg)
# Step 3.1: Replace geneID column in the original dataframe
kegg_enrichment_up_df <- as.data.frame(kegg_enrichment_up)
# Remove old geneID column and merge new one
kegg_enrichment_up_df <- kegg_enrichment_up_df %>%
dplyr::select(-geneID) %>% # Remove old geneID column
left_join(expanded_kegg %>% dplyr::select(ID, GeneID), by = "ID") %>% # Merge new GeneID column
dplyr::rename(geneID = GeneID) # Rename column back to geneID
# -----------------------------------------------------------
# ---- Perform KEGG enrichment analysis (down_regulated) ----
# Step 1: Extract KEGG KO terms from down-regulated genes
gene_list_kegg_down <- down_regulated$KEGG_ko
gene_list_kegg_down <- gsub("ko:", "", gene_list_kegg_down)
# Step 2: Perform KEGG enrichment analysis
kegg_enrichment_down <- enrichKEGG(gene = gene_list_kegg_down, organism = 'ko')
# --- Convert KEGG gene IDs (Kxxxxxx) to actual GeneIDs ---
# Step 3: Create KEGG to GeneID mapping from down_regulated dataset
kegg_to_geneid_down <- down_regulated %>%
dplyr::select(KEGG_ko, GeneID) %>%
filter(!is.na(KEGG_ko)) %>% # Remove missing KEGG KO entries
mutate(KEGG_ko = str_remove(KEGG_ko, "ko:")) # Remove 'ko:' prefix if present
# Step 4: Clean KEGG_ko values (handle multiple KEGG IDs)
kegg_to_geneid_down_clean <- kegg_to_geneid_down %>%
mutate(KEGG_ko = str_remove_all(KEGG_ko, "ko:")) %>% # Remove 'ko:' prefixes
separate_rows(KEGG_ko, sep = ",") %>% # Ensure each KEGG ID is on its own row
filter(KEGG_ko != "-") %>% # Remove invalid KEGG IDs ("-")
distinct() # Remove duplicate mappings
# Step 5: Expand geneID column in kegg_enrichment_down
expanded_kegg_down <- kegg_enrichment_down %>%
as.data.frame() %>%
separate_rows(geneID, sep = "/") %>% # Split multiple KEGG IDs (Kxxxxx)
left_join(kegg_to_geneid_down_clean, by = c("geneID" = "KEGG_ko"), relationship = "many-to-many") %>% # Handle many-to-many mappings
distinct() %>% # Remove duplicate matches
group_by(ID) %>%
summarise(across(everything(), ~ paste(unique(na.omit(.)), collapse = "/")), .groups = "drop") # Re-collapse results
# Step 6: Replace geneID column in the original kegg_enrichment_down dataframe
kegg_enrichment_down_df <- as.data.frame(kegg_enrichment_down) %>%
dplyr::select(-geneID) %>% # Remove old geneID column
left_join(expanded_kegg_down %>% dplyr::select(ID, GeneID), by = "ID") %>% # Merge new GeneID column
dplyr::rename(geneID = GeneID) # Rename column back to geneID
# View the updated dataframe
head(kegg_enrichment_down_df)
# Create a new workbook
wb <- createWorkbook()
# Save enrichment results to the workbook
addWorksheet(wb, "KEGG_Enrichment_Up")
writeData(wb, "KEGG_Enrichment_Up", as.data.frame(kegg_enrichment_up_df))
# Save enrichment results to the workbook
addWorksheet(wb, "KEGG_Enrichment_Down")
writeData(wb, "KEGG_Enrichment_Down", as.data.frame(kegg_enrichment_down_df))
#saveWorkbook(wb, "KEGG_Enrichment.xlsx", overwrite = TRUE)
# ----------------------------------------
# ---- Perform GO enrichment analysis ----
# Define gene list (up-regulated genes)
gene_list_go_up <- up_regulated$GeneID # Extract the 149 up-regulated genes
gene_list_go_down <- down_regulated$GeneID # Extract the 65 down-regulated genes
# Define background gene set (all genes in res)
background_genes <- res_updated$GeneID # Extract the 3646 background genes
# Prepare GO annotation data from res
go_annotation <- res_updated[, c("GOs","GeneID")] # Extract relevant columns
go_annotation <- go_annotation %>%
tidyr::separate_rows(GOs, sep = ",") # Split multiple GO terms into separate rows
# Perform GO enrichment analysis, where pAdjustMethod is one of "holm", "hochberg", "hommel", "bonferroni", "BH", "BY", "fdr", "none"
go_enrichment_up <- enricher(
gene = gene_list_go_up, # Up-regulated genes
TERM2GENE = go_annotation, # Custom GO annotation
pvalueCutoff = 0.05, # Significance threshold
pAdjustMethod = "BH",
universe = background_genes # Define the background gene set
)
go_enrichment_up <- as.data.frame(go_enrichment_up)
go_enrichment_down <- enricher(
gene = gene_list_go_down, # Up-regulated genes
TERM2GENE = go_annotation, # Custom GO annotation
pvalueCutoff = 0.05, # Significance threshold
pAdjustMethod = "BH",
universe = background_genes # Define the background gene set
)
go_enrichment_down <- as.data.frame(go_enrichment_down)
## Remove the 'p.adjust' column since no adjusted methods have been applied!
#go_enrichment_up <- go_enrichment_up[, !names(go_enrichment_up) %in% "p.adjust"]
# Update the Description column with the term descriptions
go_enrichment_up$Description <- sapply(go_enrichment_up$ID, function(go_id) {
# Using select to get the term description
term <- tryCatch({
AnnotationDbi::select(GO.db, keys = go_id, columns = "TERM", keytype = "GOID")
}, error = function(e) {
message(paste("Error for GO term:", go_id)) # Print which GO ID caused the error
return(data.frame(TERM = NA)) # In case of error, return NA
})
if (nrow(term) > 0) {
return(term$TERM)
} else {
return(NA) # If no description found, return NA
}
})
## Print the updated data frame
#print(go_enrichment_up)
## Remove the 'p.adjust' column since no adjusted methods have been applied!
#go_enrichment_down <- go_enrichment_down[, !names(go_enrichment_down) %in% "p.adjust"]
# Update the Description column with the term descriptions
go_enrichment_down$Description <- sapply(go_enrichment_down$ID, function(go_id) {
# Using select to get the term description
term <- tryCatch({
AnnotationDbi::select(GO.db, keys = go_id, columns = "TERM", keytype = "GOID")
}, error = function(e) {
message(paste("Error for GO term:", go_id)) # Print which GO ID caused the error
return(data.frame(TERM = NA)) # In case of error, return NA
})
if (nrow(term) > 0) {
return(term$TERM)
} else {
return(NA) # If no description found, return NA
}
})
addWorksheet(wb, "GO_Enrichment_Up")
writeData(wb, "GO_Enrichment_Up", as.data.frame(go_enrichment_up))
addWorksheet(wb, "GO_Enrichment_Down")
writeData(wb, "GO_Enrichment_Down", as.data.frame(go_enrichment_down))
# Save the workbook with enrichment results
saveWorkbook(wb, "KEGG_and_GO_Enrichments_09_MH_vs_AUM_AZI.xlsx", overwrite = TRUE)
8.3. Finalizing the KEGG and GO Enrichment table
1. NOTE (Already realized in the code): geneIDs in KEGG_Enrichment have been already translated from ko to geneID in H0N29_*-format; If not, nachmachen using eggnog-res, 因为 eggnog里有1-1-mspping Info between ko-Name and GeneID.
2. NEED_MANUAL_DELETION (Already setting the cutoff in the code): p.adjust values have been calculated, we have to filter all records in GO_Enrichment-results by |p.adjust|<=0.05. DON'T_NEED_ANY_MORE, since pvalueCutoff = 0.05 settings in enricher. Alternative using pvalueCutoff=1.0, marked the color as yellow if the p.adjusted <= 0.05 in GO_enrichment.
3. NOTE (Not occuring in the new dataset): In rare case, the description is missing for some IDs, e.g. GO term: GO:0006807: replace GO:0006807 obsolete nitrogen compound metabolic process; ko00975: Metabolism, Biosynthesis of other secondary metabolites