-
Input data:
mkdir bacto; cd bacto;
mkdir raw_data; cd raw_data;
# ── W1-4 and Y1-4 ──
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J001/01.RawData/W/W_1.fq.gz W1_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J001/01.RawData/W/W_2.fq.gz W1_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/W2/W2_1.fq.gz W2_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/W2/W2_2.fq.gz W2_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/W3/W3_1.fq.gz W3_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/W3/W3_2.fq.gz W3_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/W4/W4_1.fq.gz W4_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/W4/W4_2.fq.gz W4_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J001/01.RawData/Y/Y_1.fq.gz Y1_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J001/01.RawData/Y/Y_2.fq.gz Y1_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/Y2/Y2_1.fq.gz Y2_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/Y2/Y2_2.fq.gz Y2_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/Y3/Y3_1.fq.gz Y3_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/Y3/Y3_2.fq.gz Y3_R2.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/Y4/Y4_1.fq.gz Y4_R1.fastq.gz
ln -s ../../short/RSMD00304/X101SC24065637-Z01/X101SC24065637-Z01-J002/01.RawData/Y4/Y4_2.fq.gz Y4_R2.fastq.gz
-
Call variant calling using snippy
ln -s ~/Tools/bacto/db/ .;
ln -s ~/Tools/bacto/envs/ .;
ln -s ~/Tools/bacto/local/ .;
cp ~/Tools/bacto/Snakefile .;
cp ~/Tools/bacto/bacto-0.1.json .;
cp ~/Tools/bacto/cluster.json .;
#download CP059040.gb from GenBank
#mv ~/Downloads/sequence\(2\).gb db/CP059040.gb
mamba activate /home/jhuang/miniconda3/envs/bengal3_ac3
(bengal3_ac3) jhuang@WS-2290C:~/DATA/Data_Tam_DNAseq_2023_A6WT_A10CraA_A12AYE_A1917978$ which snakemake
/home/jhuang/miniconda3/envs/bengal3_ac3/bin/snakemake
(bengal3_ac3) jhuang@WS-2290C:~/DATA/Data_Tam_DNAseq_2023_A6WT_A10CraA_A12AYE_A1917978$ snakemake -v
4.0.0 --> CORRECT!
#NOTE_1: modify bacto-0.1.json keeping only steps assembly, typing_mlst, possibly pangenome and variants_calling true; setting cpu=20 in all used steps.
#setting the following in bacto-0.1.json
"fastqc": false,
"taxonomic_classifier": false,
"assembly": true,
"typing_ariba": false,
"typing_mlst": true,
"pangenome": true,
"variants_calling": true,
"phylogeny_fasttree": false,
"phylogeny_raxml": false,
"recombination": false, (due to gubbins-error set false)
"prokka": {
"genus": "Acinetobacter",
"kingdom": "Bacteria",
"species": "baumannii",
"cpu": 10,
"evalue": "1e-06",
"other": ""
},
"mykrobe": {
"species": "abaum"
},
"reference": "db/CP059040.gb"
#NOTE_2: needs disk Titisee since the pipeline needs /media/jhuang/Titisee/GAMOLA2/TIGRfam_db/TIGRFAMs_15.0_HMM.LIB
snakemake --printshellcmds
-
Summarize all SNPs and Indels from the snippy result directory.
cp ~/Scripts/summarize_snippy_res_ordered.py .
# IMPORTANT_ADAPT the array in script should be adapted
isolates = ["W1", "W2", "W3", "W4", "Y1", "Y2", "Y3", "Y4"]
mamba activate plot-numpy1
python3 ./summarize_snippy_res_ordered.py snippy
#--> Summary CSV file created successfully at: snippy/summary_snps_indels.csv
cd snippy
#REMOVE_the_line? I don't find the sence of the line: grep -v "None,,,,,,None,None" summary_snps_indels.csv > summary_snps_indels_.csv
-
Using spandx calling variants (almost the same results to the one from viral-ngs!)
mamba deactivate
mamba activate /home/jhuang/miniconda3/envs/spandx
# PREPARE the inputs for the options ref and database
mkdir ~/miniconda3/envs/spandx/share/snpeff-5.1-2/data/PP810610
cp PP810610.gb ~/miniconda3/envs/spandx/share/snpeff-5.1-2/data/PP810610/genes.gbk
vim ~/miniconda3/envs/spandx/share/snpeff-5.1-2/snpEff.config
/home/jhuang/miniconda3/envs/spandx/bin/snpEff build PP810610 #-d
~/Scripts/genbank2fasta.py PP810610.gb
mv PP810610.gb_converted.fna PP810610.fasta #rename "NC_001348.1 xxxxx" to "NC_001348" in the fasta-file
ln -s /home/jhuang/Tools/spandx/ spandx
(spandx) nextflow run spandx/main.nf --fastq "trimmed/*_P_{1,2}.fastq" --ref CP059040.fasta --annotation --database CP059040 -resume
# RERUN SNP_matrix.sh due to the error ERROR_CHROMOSOME_NOT_FOUND in the variants annotation, resulting in all impacts are MODIFIER --> IT WORKS!
cd Outputs/Master_vcf
conda activate /home/jhuang/miniconda3/envs/spandx
(spandx) cp -r ../../snippy/Y1/reference . # Eigentlich irgendein directory, all directories contains the same reference.
(spandx) cp ../../spandx/bin/SNP_matrix.sh ./
#Note that ${variant_genome_path}=CP059040 in the following command, but it was not used after the following command modification.
#Adapt "snpEff eff -no-downstream -no-intergenic -ud 100 -formatEff -v ${variant_genome_path} out.vcf > out.annotated.vcf" to
"/home/jhuang/miniconda3/envs/bengal3_ac3/bin/snpEff eff -no-downstream -no-intergenic -ud 100 -formatEff -c reference/snpeff.config -dataDir . ref out.vcf > out.annotated.vcf" in SNP_matrix.sh
(spandx) bash SNP_matrix.sh CP059040 .
-
Calling inter-host variants by merging the results from snippy+spandx
Cross-Caller SNP/Indel Concordance & Invariant Variant Analyzer;
Multi-Isolate Variant Intersection, Annotation Harmonization & Caller Discrepancy Report;
Comparative Genomic Variant Profiling: Concordance, Invariance & Allele Mismatch Analysis;
VarMatch: Cross-Tool Variant Concordance Pipeline
mamba activate plot-numpy1
cd bacto
cp Outputs/Master_vcf/All_SNPs_indels_annotated.txt .
cp snippy/summary_snps_indels.csv .
cp ~/Scripts/process_variants_snippy_alleles_spandx_annotations.py .
#Configuring
ISOLATES = [
"Y1", "Y2", "Y3", "Y4", "W1", "W2", "W3", "W4"
]
(plot-numpy1) python process_variants_snippy_alleles_spandx_annotations.py
# mv common_variants_all_snippy_annotated.xlsx common_variants_snippy+spandx_annotated_Y1Y2Y3Y4W1W2W3W4.xlsx
# mv common_variants_invariant_snippy_annotated.xlsx common_invariants_snippy+spandx_annotated_Y1Y2Y3Y4W1W2W3W4.xlsx
-
Manully checking each of the 6 records by comparing them to the results from SPANDx; three are confirmed!
#CHROM,POS,REF,ALT,TYPE,Y1,Y2,Y3,Y4,W1,W2,W3,W4,Effect,Impact,Functional_Class,Codon_change,Protein_and_nucleotide_change,Amino_Acid_Length,Gene_name,Biotype
# -- Results from snippy --
#move: CP059040,1527276,TTGAACC,T,del,TTGAACC,TTGAACC,TTGAACC,T,TTGAACC,TTGAACC,T,T,conservative_inframe_deletion,MODERATE,,gaacct/,p.Glu443_Pro444del/c.1327_1332delGAACCT,704,H0N29_07175,protein_coding
#confirmed: CP059040,1843289,G,T,snp,G,T,G,G,G,G,G,G,missense_variant,MODERATE,MISSENSE,gCg/gAg,p.Ala37Glu/c.110C>A,357,H0N29_08665,protein_coding
#confirmed: CP059040,2019186,G,A,snp,A,G,G,G,G,G,G,G,upstream_gene_variant,MODIFIER,,59,c.-59C>T,144,H0N29_09480,protein_coding
#delete_this? CP059040,3124917,T,"TAC,TACTTCATTACATACCAACCGCCAAGGGTGC",snp,C,T,C,C,T,T,T,C,upstream_gene_variant,MODIFIER,,25,c.-25_-24insAC,0,H0N29_00075,protein_coding
#move: CP059040,3310021,C,CT,ins,CT,CT,CT,CT,C,CT,CT,CT,intragenic_variant,MODIFIER,,,n.3310021_3310022insT,,H0N29_00075,
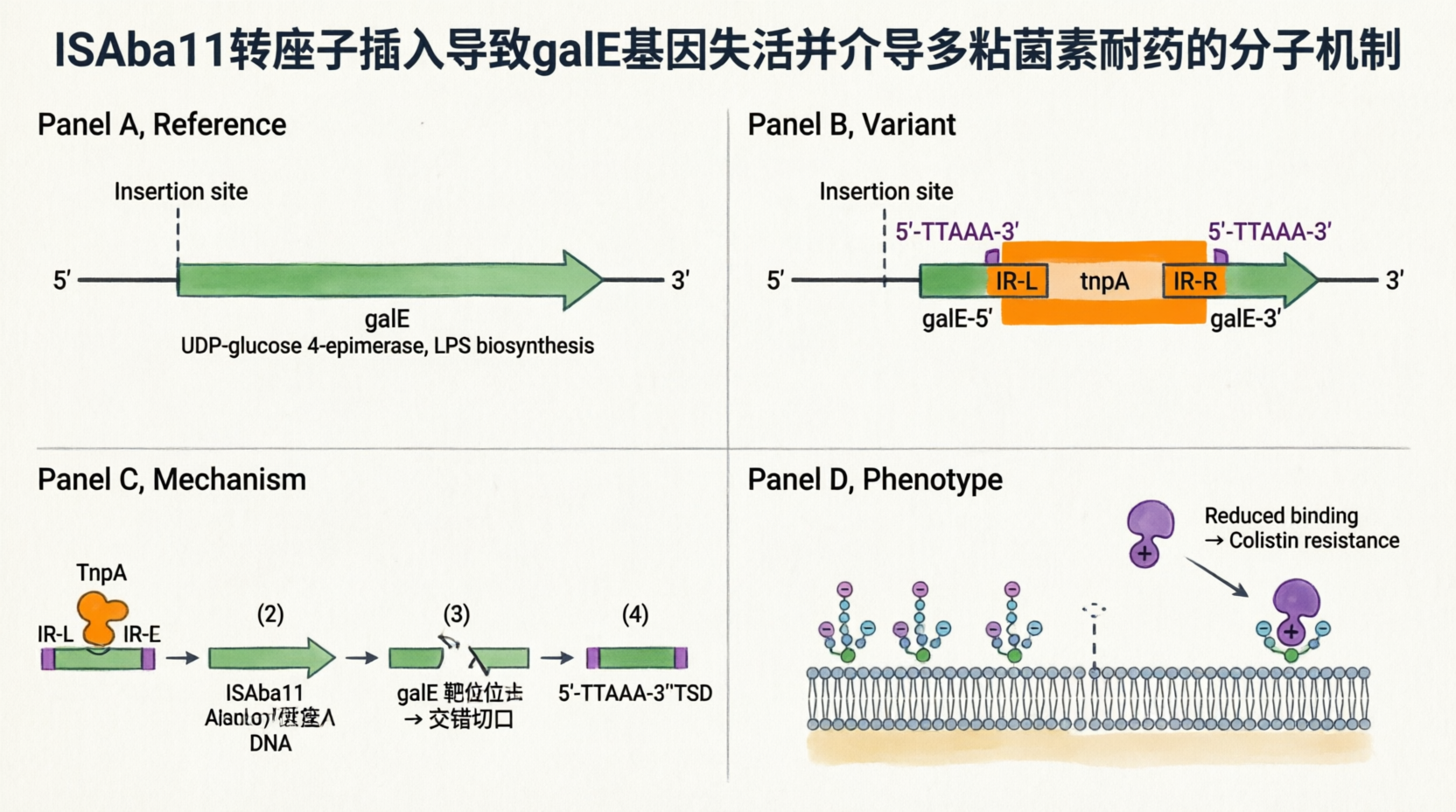
#confirmed: CP059040,3853714,G,A,snp,G,G,G,G,G,A,G,A,stop_gained,HIGH,NONSENSE,Cag/Tag,p.Gln91*/c.271C>T,338,H0N29_18290,protein_coding
#--> Only three SNPs are confirmed --> means there is almost no variation in the genomic level!
# -- Results from the SPANDx --
#CP059040 1527276 TTGAACC T INDEL TTGAACC/T T T T T T T T conservative_inframe_deletion MODERATE gaacct/ p.Glu443_Pro444del/c.1327_1332delGAACCT 704 H0N29_07175 protein_coding
#CP059040 1843289 G T SNP G T G G G G G G missense_variant MODERATE MISSENSE gCg/gAg p.Ala37Glu/c.110C>A 357 H0N29_08665 protein_coding
#CP059040 2019186 G A SNP A G G G G G G G upstream_gene_variant MODIFIER 59 c.-59C>T 144 H0N29_09480 protein_coding
#Cmp to CP059040 3124917 T TAC,TACTTCATTACATACCAACCGCCAAGGGTGC INDEL . TACTTCATTACATACCAACCGCCAAGGGTGC TACTTCATTACATACCAACCGCCAAGGGTGC TAC . . . . upstream_gene_variant MODIFIER 25 c.-25_-24insAC 0 H0N29_00075 protein_coding
#Cmp to CP059040 3124920 C CATTACATACCAACCGCCAAGGGTGCTTCATG INDEL . . . CATTACATACCAACCGCCAAGGGTGCTTCATG . . C . upstream_gene_variant MODIFIER 22 c.-22_-21insATTACATACCAACCGCCAAGGGTGCTTCATG 0 H0N29_00075 protein_coding
#TODO: Move to invariant-file: CP059040 3310021 C CT INDEL CT CT CT CT CT CT CT CT intragenic_variant MODIFIER n.3310021_3310022insT H0N29_00075
#CP059040 3853714 G A SNP G G G G G A G A stop_gained HIGH NONSENSE Cag/Tag p.Gln91*/c.271C>T 338 H0N29_18290 protein_coding
#-->For the Indel-report, more complicated, needs the following command to find the initial change and related codon-change.
## Check gene strand in your GFF/GenBank
#grep "H0N29_07175" reference.gff
# Extract 20 bp around the variant from reference
samtools faidx CP059040.fasta CP059040:1527260-1527290
-
Annotation of the three confirmed SNPs
gene complement(3852968..3853984)
/gene="galE"
/locus_tag="H0N29_18290"
CDS complement(3852968..3853984)
/gene="galE"
/locus_tag="H0N29_18290"
/EC_number="5.1.3.2"
/inference="COORDINATES: similar to AA
sequence:RefSeq:WP_017725586.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="UDP-glucose 4-epimerase GalE"
/protein_id="QNT84923.1"
/translation="MAKILVTGGAGYIGSHTCVELLEAGHEVIVFDNLSNSSKESLNR
VQEITQKGLTFVEGDIRNSGELDRVFQEHAIDAVIHFAGLKAVGESQEKPLIYFDNNI
AGSIQLVKSMEKAGVYTLVFSSSATVYDEANTSPLNEEMPTGMPSNNYGYTKLIVEQL
LQKLSVADSKWSIALLRYFNPVGAHKSGRIGEDPQGIPNNLMPYVTQVAVGRREKLSI
YGNDYDTIDGTGVRDYIHVVDLANAHLCALNNRLQAQGCRAWNIGTGNGSSVLQVKNT
FEQVNGVPVAFEFAPRRAGDVATSFADNARAVAELGWKPQYGLEDMLKDSWNWQKQNP
NGYN"
gene complement(1842325..1843398)
/gene="adeS"
/locus_tag="H0N29_08665"
CDS complement(1842325..1843398)
/gene="adeS"
/locus_tag="H0N29_08665"
/inference="COORDINATES: similar to AA
sequence:RefSeq:WP_000837467.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="two-component sensor histidine kinase AdeS"
/protein_id="QNT86623.1"
/translation="MKSKLGISKQLFIALTIVNLSVTLFSVVLGYVIYNYAIEKGWIS
LSSFQQEDWTSFHFVDWIWLATVIFCGCIISLVIGMRLAKRFIVPINFLAEAAKKISH
GDLSARAYDNRIHSAEMSELLYNFNDMAQKLEVSVKNAQVWNAAIAHELRAPITILQG
RLQGIIDGVFKPDEVLFKSLLNQVEGLSHLVEDLRTLSLVENQQLRLNYELFDLKAVV
EKVLKAFEDRLDQAKLVPELDLTSTPVYCDRRRIEQVLIALIDNSIRYSNAGKLKISS
EVVADNWILKIEDEGPGIATEFQDDLFKPFFRLEESRNKEFGGTGLGLAVVHAIVVAL
KGTIQYSNQGSKSVFTIKISMNN"
gene complement(2018693..2019127)
/locus_tag="H0N29_09480"
CDS complement(2018693..2019127)
/locus_tag="H0N29_09480"
/inference="COORDINATES: similar to AA
sequence:RefSeq:YP_004995263.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="HIT domain-containing protein"
/protein_id="QNT83319.1"
/translation="MFSLHPQLAQDTFFVGDFPLSTCRLMNDMQFPWLILVPRVPGIT
ELYELSQADQEQFLRESSWLSSQLSRVFRADKMNVAALGNMVPQLHFHHVVRYQNDVA
WPKPVWGTPAVPYTSDVLAHMRQTLMLALRGQGDMPFDWRMD"
-
Structural Variant Detection
conda activate sv_assembly
# Step 1: Align assemblies to reference
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./Y1_unicycler_out/assembly.fasta -p Y1
# Step 2: Filter alignments (1-to-1 best matches)
delta-filter -1 -q Y1.delta > Y1.filtered.delta
# Note: Use -1 for 1-to-1, not -r -q
# Step 3: Run Assemblytics with ALL 5 parameters
Assemblytics Y1.filtered.delta Y1_assemblytics 1000 100 50000
# Step 5: Extract large insertions only
grep -w "Insertion" Y1_assemblytics.Assemblytics_structural_variants.bed > Y1_insertions.bed
# 6. Check if ANY variants were detected (any size)
wc -l Y1_assemblytics.Assemblytics_structural_variants.bed
# 7. View variant type distribution
cut -f4 Y1_assemblytics.Assemblytics_structural_variants.bed | sort | uniq -c
# 8. Check alignment coverage (are contigs aligning well?)
cat Y1_assemblytics.Assemblytics_assembly_stats.txt
# 9. Check raw delta file for alignment blocks
show-coords -rcl Y1.filtered.delta | head -20
# 10. If bed file is empty, try relaxing parameters and re-run:
Assemblytics Y1.filtered.delta Y1_assemblytics_v2 500 50 100000
# └─unique─┘ └min┘ └──max──┘
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./Y1_unicycler_out/assembly.fasta -p Y1
delta-filter -1 -q Y1.delta > Y1.filtered.delta
Assemblytics Y1.filtered.delta Y1_assemblytics 1000 100 50000
vim Y1_assemblytics.variant_preview.txt
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
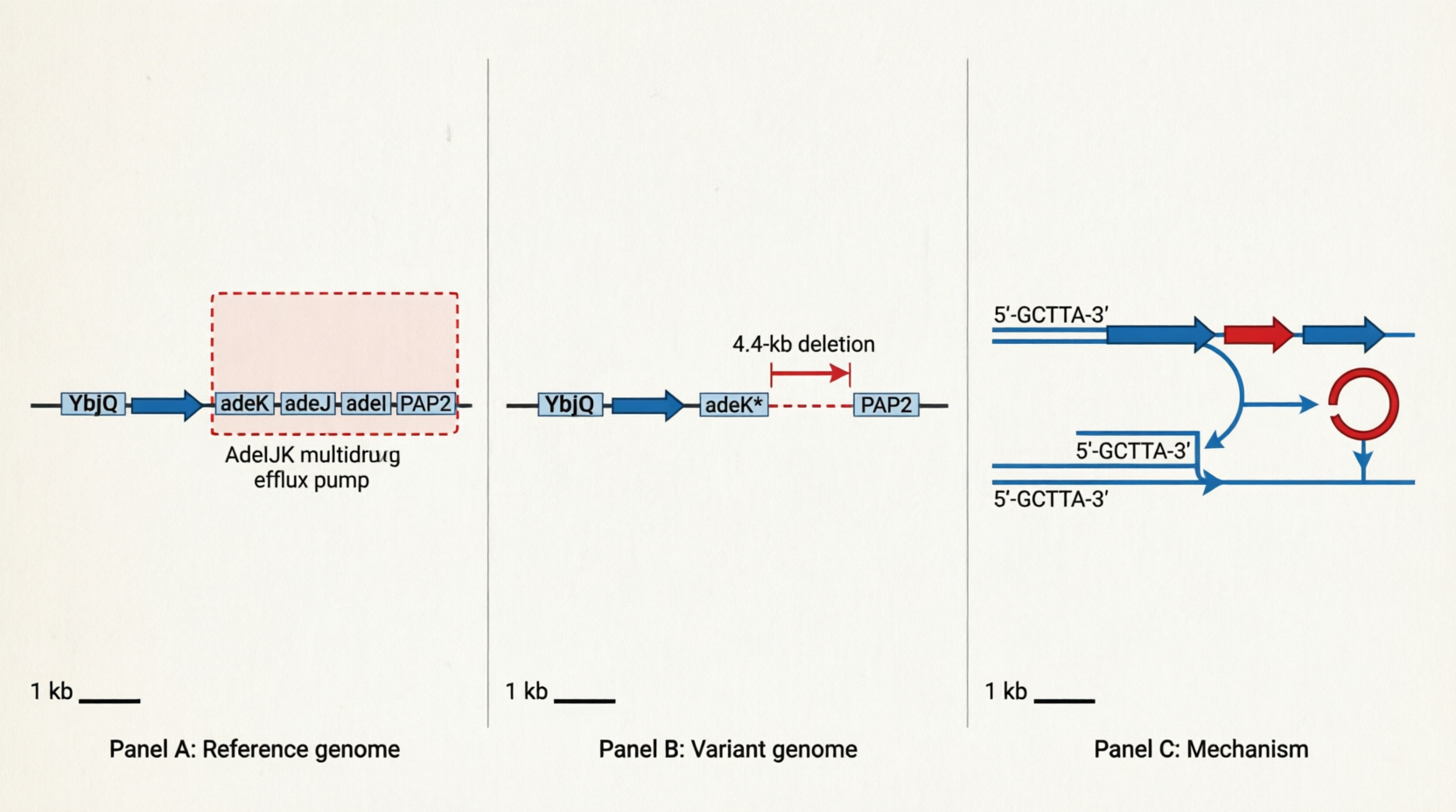
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737272-737279:+ between_alignments
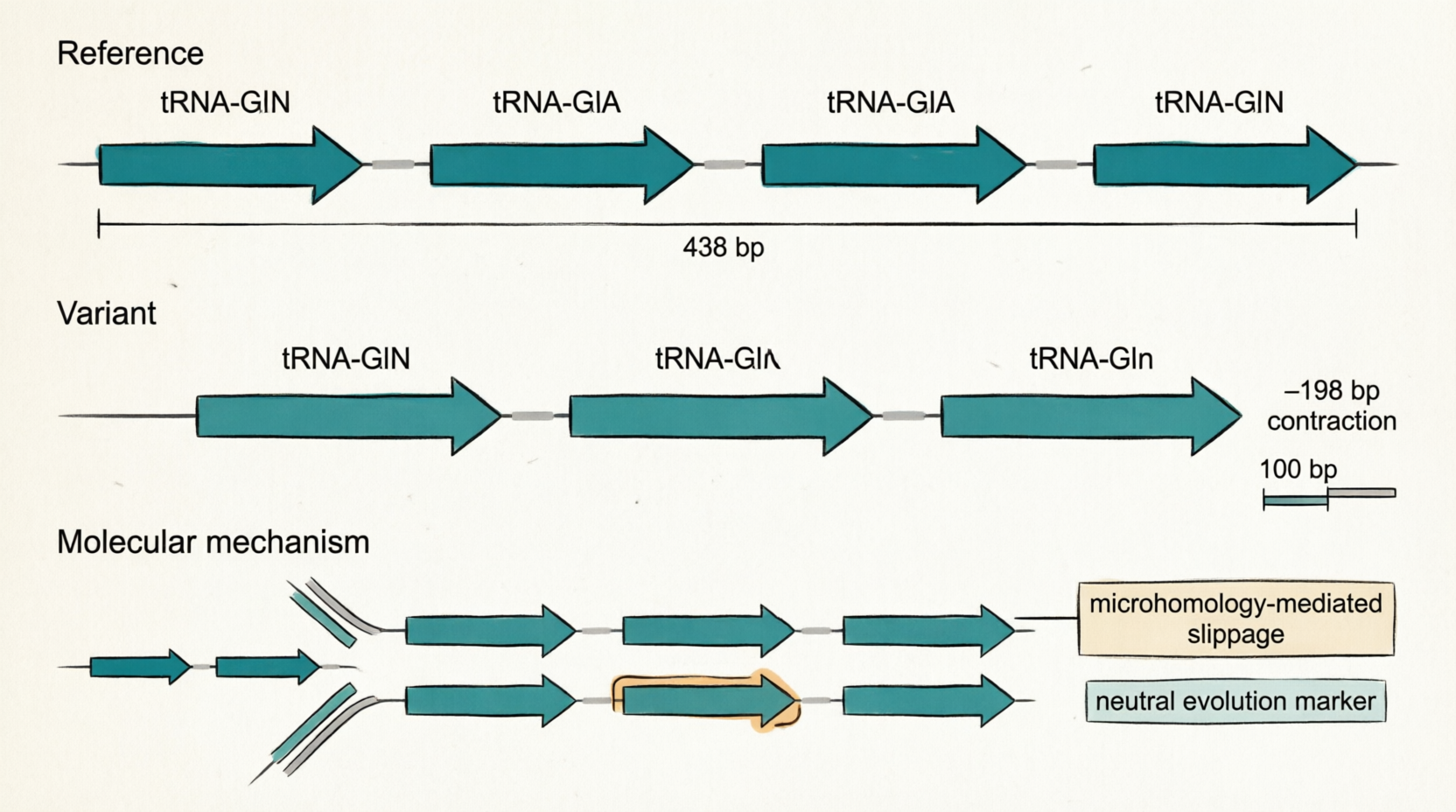
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067782-3067859:- between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./Y2_unicycler_out/assembly.fasta -p Y2
delta-filter -1 -q Y2.delta > Y2.filtered.delta
Assemblytics Y2.filtered.delta Y2_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737272-737279:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067782-3067859:- between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./Y3_unicycler_out/assembly.fasta -p Y3
delta-filter -1 -q Y3.delta > Y3.filtered.delta
Assemblytics Y3.filtered.delta Y3_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737281-737288:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067791-3067868:- between_alignments
CP059040 3439419 3439424 Assemblytics_b_6 1106 + Insertion -5 1101 1:3382176-3383277:+ between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./Y4_unicycler_out/assembly.fasta -p Y4
delta-filter -1 -q Y4.delta > Y4.filtered.delta
Assemblytics Y4.filtered.delta Y4_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 cluster_001_consensus:737272-737279:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 cluster_001_consensus:3067776-3067853:- between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./W1_unicycler_out/assembly.fasta -p W1
delta-filter -1 -q W1.delta > W1.filtered.delta
Assemblytics W1.filtered.delta W1_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737272-737279:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067782-3067859:- between_alignments
CP059040 3853883 3853888 Assemblytics_b_6 1106 + Insertion -5 1101 1:3796629-3797730:+ between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./W2_unicycler_out/assembly.fasta -p W2
delta-filter -1 -q W2.delta > W2.filtered.delta
Assemblytics W2.filtered.delta W2_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737272-737279:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067782-3067859:- between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./W3_unicycler_out/assembly.fasta -p W3
delta-filter -1 -q W3.delta > W3.filtered.delta
Assemblytics W3.filtered.delta W3_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737272-737279:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067782-3067859:- between_alignments
CP059040 3853884 3853890 Assemblytics_b_6 1106 + Insertion -6 1100 1:3796631-3797731:+ between_alignments
nucmer --maxmatch -l 100 -c 500 bacto/CP059040.fasta ./W4_unicycler_out/assembly.fasta -p W4
delta-filter -1 -q W4.delta > W4.filtered.delta
Assemblytics W4.filtered.delta W4_assemblytics 1000 100 50000
#reference ref_start ref_stop ID size strand type ref_gap_size query_gap_size query_coordinates method
CP059040 737224 741667 Assemblytics_b_2 4436 + Deletion 4443 7 1:737272-737279:+ between_alignments
CP059040 3124916 3125037 Assemblytics_b_5 198 + Tandem_contraction 121 -77 1:3067782-3067859:- between_alignments
samtools faidx ./W1_unicycler_out/assembly.fasta 1:3796629-3797730
samtools faidx ./W3_unicycler_out/assembly.fasta 1:3796631-3797731
gene 3124675..3124749
/locus_tag="H0N29_14850"
tRNA 3124675..3124749
/locus_tag="H0N29_14850"
/product="tRNA-Gln"
/inference="COORDINATES: profile:tRNAscan-SE:2.0.4"
/note="Derived by automated computational analysis using
gene prediction method: tRNAscan-SE."
/anticodon=(pos:3124707..3124709,aa:Gln,seq:ttg)
gene 3124841..3124915
/locus_tag="H0N29_14855"
tRNA 3124841..3124915
/locus_tag="H0N29_14855"
/product="tRNA-Gln"
/inference="COORDINATES: profile:tRNAscan-SE:2.0.4"
/note="Derived by automated computational analysis using
gene prediction method: tRNAscan-SE."
/anticodon=(pos:3124873..3124875,aa:Gln,seq:ttg)
gene 3124943..3125017
/locus_tag="H0N29_14860"
tRNA 3124943..3125017
/locus_tag="H0N29_14860"
/product="tRNA-Gln"
/inference="COORDINATES: profile:tRNAscan-SE:2.0.4"
/note="Derived by automated computational analysis using
gene prediction method: tRNAscan-SE."
/anticodon=(pos:3124975..3124977,aa:Gln,seq:ttg)
gene 3125039..3125113
/locus_tag="H0N29_14865"
tRNA 3125039..3125113
/locus_tag="H0N29_14865"
/product="tRNA-Gln"
/inference="COORDINATES: profile:tRNAscan-SE:2.0.4"
/note="Derived by automated computational analysis using
gene prediction method: tRNAscan-SE."
/anticodon=(pos:3125071..3125073,aa:Gln,seq:ttg)
#--
gene complement(735319..735669)
/locus_tag="H0N29_03535"
CDS complement(735319..735669)
/locus_tag="H0N29_03535"
/inference="COORDINATES: similar to AA
sequence:RefSeq:YP_004996456.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="YbjQ family protein"
/protein_id="QNT85684.1"
/db_xref="GI:1906909114"
/translation="MLLSNLESVPGHQILKQLDVVYGSTVRSKHVGRDLMASLKNIVG
GELTGYTELLEESRQEAMQRMIAKAEQLGANAIVGIRFSTSNIAQGASELFVYGTAVV
VQPQAPHLPDPFNA"
gene complement(735779..737233)
/gene="adeK"
/locus_tag="H0N29_03540"
CDS complement(735779..737233)
/gene="adeK"
/locus_tag="H0N29_03540"
/inference="COORDINATES: similar to AA
sequence:RefSeq:YP_004996455.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="multidrug efflux RND transporter outer membrane
channel subunit AdeK"
/protein_id="QNT86781.1"
/db_xref="GI:1906910211"
/translation="MQKVWSISGRSIAVSALALALAACQSMRGPEPVVKTDIPQSYAY
NSASGTSIAEQGYKQFFADPRLLEVIDLALANNRDLRTATLNIERAQQQYQITQNNQL
PTIGASGSAIRQVSQSRDPNNPYSTYQVGLGVTAYELDFWGRVRSLKDAALDSYLATQ
SARDSTQISLISQVAQAWLNYSFATANLRLAEQTLKAQLDSYNLNKKRFDVGIDSEVP
LRQAQISVETARNDVANYKTQIAQAQNLLNLLVGQPVPQNLLPTQPVKRIAQQNVFTA
GLPSDLLNNRPDVKAAEYNLSAAGANIGAAKARLFPTISLTGSAGYASTDLSDLFKSG
GFVWSVGPSLDLPIFDWGTRRANVKISETDQKIALSDYEKSVQSAFREVNDALATRAN
IGERLTAQQRLVEATNRNYTLSNARFRAGIDSYLTVLDAQRSSYAAEQGLLLLQQANL
NNQIELYKTLGGGLKANTSDTVVHQPSSAELKKQ"
gene complement(737233..740409)
/gene="adeJ"
/locus_tag="H0N29_03545"
CDS complement(737233..740409)
/gene="adeJ"
/locus_tag="H0N29_03545"
/inference="COORDINATES: similar to AA
sequence:RefSeq:WP_116497130.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="multidrug efflux RND transporter permease
subunit AdeJ"
/protein_id="QNT85685.1"
/db_xref="GI:1906909115"
/translation="MAQFFIHRPIFAWVIALVIMLAGILTLTKMPIAQYPTIAPPTVT
IAATYPGASAETVENTVTQIIEQQMNGLDGLRYISSNSAGNGQASIQLNFEQGVDPDI
AQVQVQNKLQSATALLPEDVQRQGVTVTKSGASFLQVIAFYSPDNNLSDSDIKDYVNS
SIKEPLSRVAGVGEVQVFGGSYAMRIWLDPAKLTSYQLTPSDIATALQAQNSQVAVGQ
LGGAPAVQGQVLNATVNAQSLLQTPEQFKNIFLKNTASGAEVRLKDVARVELGSDNYQ
FDSKFNGKPAAGLAIKIATGANALDTAEAVEQRLSELRKNYPTGLADKLAYDTTPFIR
LSIESVVHTLIEAVILVFIVMFLFLQNWRATIIPTLAVPVVVLGTFAVINIFGFSINT
LTMFAMVLAIGLLVDDAIVVVENVERVMSEDHTDPVTATSRSMQQISGALVGITSVLT
AVFVPMAFFGGTTGVIYRQFSITLVTAMVLSLIVALTFTPALCATILKQHDPNKEPSN
NIFARFFRSFNNGFDRMSHSYQNGVSRMLKGKIFSGVLYAVVVALLVFLFQKLPSSFL
PEEDQGVVMTLVQLPPNATLDRTGKVIDTMTNFFMNEKDTVESIFTVSGFSFTGVGQN
AGIGFVKLKDWSKRTTPETQIGSLIQRGMALNMIIKDASYVMPLQLPAMPELGVTAGF
NLQLKDSSGQGHEKLIAARNTILGLASQDKRLVGVRPNGQEDTPQYQINVDQAQAGAM
GVSIAEINNTMRIAWGGSYINDFVDRGRVKKVYVQGDAGSRMMPEDLNKWYVRNNKGE
MVPFSAFATGEWTYGSPRLERYNGVSSVNIQGTPAPGVSSGDAMKAMEEIIGKLPSMG
LQGFDYEWTGLSLEERESGAQAPFLYALSLLIVFLCLAALYESWSIPFSVLLVVPLGV
IGAIVLTYLGMIIKGDPNLSNNIYFQVAIIAVIGLSAKNAILIVEFAKELQEKGEDLL
DATLHAAKMRLRPIIMTTLAFGFGVLPLALSTGAGAGSQHSVGFGVLGGVLSATFLGI
FFIPVFYVWIRSIFKYKPKTINTQEHKS"
gene complement(740422..741672)
/gene="adeI"
/locus_tag="H0N29_03550"
CDS complement(740422..741672)
/gene="adeI"
/locus_tag="H0N29_03550"
/inference="COORDINATES: similar to AA
sequence:RefSeq:YP_004996452.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="multidrug efflux RND transporter periplasmic
adaptor subunit AdeI"
/protein_id="QNT85686.1"
/db_xref="GI:1906909116"
/translation="MMSAKLWAPALTACALATSIALVGCSKGSDEKQQAAAAQKMPPA
EVGVIVAQPQSVEQSVELSGRTSAYQISEVRPQTSGVILKRLFAEGSYVREGQALYEL
DSRTNRATLENAKASLLQQQANLASLRTKLNRYKQLVSSNAVSKQEYDDLLGQVNVAE
AQVAAAKAQVTNANVDLGYSTIRSPISGQSGRSSVTAGALVTANQTDPLVTIQQLDPI
YVDINQSSAELLRLRQQLSKGSLNNSNNTKVKLKLEDGSTYPIEGQLAFSDASVNQDT
GTITLRAVFSNPNHLLLPGMYTTAQIVQGVVPNAYLIPQAAITRLPTGQAVAMLVNAK
GVVESRPVETSGVQGQNWIVTNGLKAGDKVIVDGVAKVKEGQEVSAKPYQAQPANSQG
AAPNAAKPAQSGKPQAEQKAASNA"
gene complement(741697..742323)
/locus_tag="H0N29_03555"
CDS complement(741697..742323)
/locus_tag="H0N29_03555"
/inference="COORDINATES: similar to AA
sequence:RefSeq:YP_004996451.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="phosphatase PAP2 family protein"
/protein_id="QNT85687.1"
/db_xref="GI:1906909117"
/translation="MPYLLLCIGCVFLGLGVLGLFVPSLQSLDLLTVQTLSHHRLDYL
NTITTFLARVGGMPFVCFLSFLVCIYLAWYKKYITVIFISLGVIGSITMGWLLKWCVN
RPRPPEAYHIVESYGASFPSAHSVYASTLACLAMIMLCHKHNINSPYIVLISCLWFVC
MGLSRIYAGVHFPTDVLAGWGIGFIWIALLWLWLLQTQSRLSRKQIYF"
#--
gene complement(3852968..3853984)
/gene="galE"
/locus_tag="H0N29_18290"
CDS complement(3852968..3853984)
/gene="galE"
/locus_tag="H0N29_18290"
/EC_number="5.1.3.2"
/inference="COORDINATES: similar to AA
sequence:RefSeq:WP_017725586.1"
/note="Derived by automated computational analysis using
gene prediction method: Protein Homology."
/codon_start=1
/transl_table=11
/product="UDP-glucose 4-epimerase GalE"
/protein_id="QNT84923.1"
/db_xref="GI:1906908353"
/translation="MAKILVTGGAGYIGSHTCVELLEAGHEVIVFDNLSNSSKESLNR
VQEITQKGLTFVEGDIRNSGELDRVFQEHAIDAVIHFAGLKAVGESQEKPLIYFDNNI
AGSIQLVKSMEKAGVYTLVFSSSATVYDEANTSPLNEEMPTGMPSNNYGYTKLIVEQL
LQKLSVADSKWSIALLRYFNPVGAHKSGRIGEDPQGIPNNLMPYVTQVAVGRREKLSI
YGNDYDTIDGTGVRDYIHVVDLANAHLCALNNRLQAQGCRAWNIGTGNGSSVLQVKNT
FEQVNGVPVAFEFAPRRAGDVATSFADNARAVAELGWKPQYGLEDMLKDSWNWQKQNP
NGYN"
-
Reporting plasmid of the 8 samples
grep ">" ./Y1_unicycler_out/assembly.fasta
grep ">" ./Y2_unicycler_out/assembly.fasta
grep ">" ./Y3_unicycler_out/assembly.fasta
grep ">" ./Y4_unicycler_out/assembly.fasta
grep ">" ./W1_unicycler_out/assembly.fasta
grep ">" ./W2_unicycler_out/assembly.fasta
grep ">" ./W3_unicycler_out/assembly.fasta
grep ">" ./W4_unicycler_out/assembly.fasta
>1 length=3923593 depth=1.00x circular=true
>2 length=17195 depth=32.32x circular=true
>1 length=3923593 depth=1.00x circular=true
>2 length=9540 depth=41.67x circular=true
>3 length=7655 depth=35.32x circular=true
>1 length=3924708 depth=1.00x circular=true
>2 length=7655 depth=33.84x circular=true
>cluster_001_consensus 3923587 23 3923587 3923588
>cluster_004_consensus 9538 3923634 9538 9539
>cluster_008_consensus 7655 3933196 7655 7656
>1 length=3924699 depth=1.00x circular=true
>2 length=17195 depth=39.78x circular=true
>1 length=3923593 depth=1.00x circular=true
>2 length=24850 depth=31.68x circular=true
>1 length=3924699 depth=1.00x circular=true
>2 length=16750 depth=43.47x circular=true
>1 length=3923593 depth=1.00x circular=true
>2 length=9540 depth=43.37x circular=true
>3 length=7655 depth=33.45x circular=true
samtools faidx Y1_unicycler_out/assembly.fasta 2 >> plasmids/Y1_plasmids.fasta
samtools faidx Y2_unicycler_out/assembly.fasta 2 >> plasmids/Y2_plasmids.fasta
samtools faidx Y2_unicycler_out/assembly.fasta 3 >> plasmids/Y2_plasmids.fasta
samtools faidx Y3_unicycler_out/assembly.fasta 2 >> plasmids/Y3_plasmids.fasta
samtools faidx Y4_unicycler_out/assembly.fasta cluster_004_consensus >> plasmids/Y4_plasmids.fasta
samtools faidx Y4_unicycler_out/assembly.fasta cluster_008_consensus >> plasmids/Y4_plasmids.fasta
samtools faidx W1_unicycler_out/assembly.fasta 2 >> plasmids/W1_plasmids.fasta
samtools faidx W2_unicycler_out/assembly.fasta 2 >> plasmids/W2_plasmids.fasta
samtools faidx W3_unicycler_out/assembly.fasta 2 >> plasmids/W3_plasmids.fasta
samtools faidx W4_unicycler_out/assembly.fasta 2 >> plasmids/W4_plasmids.fasta
samtools faidx W4_unicycler_out/assembly.fasta 3 >> plasmids/W4_plasmids.fasta
cd plasmids
mash sketch -i *.fasta -o all_plasmids.msh
mash dist all_plasmids.msh all_plasmids.msh > mash_distances.txt
asmid,Length,Fusion Score,Verdict
Y1_17195nt,"17,195 bp",1.052,✅ True p1+p2 fusion
W1_17195nt,"17,195 bp",1.052,✅ True p1+p2 fusion (identical to Y1)
W2_24850nt,"24,850 bp",1.052,🔶 p1+p2 fusion + ~7.6 kb cargo/duplication
W3_16750nt,"16,750 bp",0.952,❌ p1-like variant", not a fusion" [deletion of 445 bp]
#How to Check if p2-Like Sequences Are Integrated in Y3's Chromosome
# If Y3 chromosome shares <100 hashes with p2, it's absent
mash dist <(mash sketch -o y3_chr Y3_unicycler_out/assembly.fasta) plasmids/p2ATCC19606.fasta | awk '$4 < 100 {print "p2 ABSENT in Y3 chromosome"}'
# Extract contig "1" (the ~3.9 Mb chromosome) from Y3 assembly
samtools faidx Y3_unicycler_out/assembly.fasta "1" > Y3_chromosome.fasta
# If you haven't sketched p2 yet:
mash sketch plasmids/p2ATCC19606.fasta -o p2_ref
# Screen Y3 chromosome against p2 (much faster than dist)
mash screen p2_ref.msh Y3_chromosome.fasta > y3_chr_vs_p2.screen
cat y3_chr_vs_p2.screen
# Extract shared hashes and check if < 100
mash screen p2_ref.msh Y3_chromosome.fasta | awk -F'\t' '{
split($5, a, "/");
if (a[1] < 100) print "✅ p2 ABSENT in Y3 chromosome (only " a[1] "/1000 k-mers shared)";
else if (a[1] > 800) print "🔴 p2 PRESENT in Y3 chromosome (" a[1] "/1000 k-mers shared)";
else print "⚠️ Partial p2 homology in Y3 chromosome (" a[1] "/1000 k-mers shared)";
}'
# Make BLAST db from Y3 chromosome
makeblastdb -in Y3_chromosome.fasta -dbtype nucl -out y3_chr_db
# Search p2 against Y3 chromosome (sensitive settings)
blastn -query plasmids/p2ATCC19606.fasta -db y3_chr_db \
-outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore" \
-evalue 1e-10 -word_size 11 -max_target_seqs 5 -out y3_chr_vs_p2.blastn
# Check results
if [ -s y3_chr_vs_p2.blastn ]; then
echo "🔍 Potential p2 integration found:"
head -5 y3_chr_vs_p2.blastn | column -t
else
echo "✅ No significant p2 homology in Y3 chromosome"
fi
Plasmid Group,Samples,Verdict,Action
~7.6 kb,"Y2, Y3, Y4, W4",🟢 No difference,Report as conserved
~9.5 kb,"Y2, Y4, W4",🟢 No difference,Report as conserved
~17 kb,"Y1, W1, W3",🟡 Moderate difference,Align to identify indel
24.8 kb,W2,🔴 Big difference,Report as unique plasmid
Plasmid,Length,Fusion Score,Verdict
Y1_17195nt,"17,195 bp",1.052,✅ True p1+p2 fusion
W1_17195nt,"17,195 bp",1.052,✅ True p1+p2 fusion (identical to Y1)
W2_24850nt,"24,850 bp",1.052,🔶 p1+p2 fusion + ~7.6 kb cargo/duplication
W3_16750nt,"16,750 bp",0.952,❌ p1-like variant", not a fusion"
But the W3_16750nt has two records mapping p1 and p2, I think they should contains all genes of p1 and p2.
Acinetobacter baumannii strain ATCC 19606 plasmid p1ATCC19606, complete sequence
Sequence ID: CP045108.1Length: 7655Number of Matches: 9
Related Information
Gene-associated gene details
Range 1: 1 to 7655GenBankGraphicsNext MatchPrevious Match
Alignment statistics for match #1
Score Expect Identities Gaps Strand
14098 bits(7634) 0.0 7649/7656(99%) 2/7656(0%) Plus/Plus
Acinetobacter baumannii strain ATCC 19606 plasmid p2ATCC19606, complete sequence
Sequence ID: CP045109.1Length: 9540Number of Matches: 8
Related Information
Gene-associated gene details
Range 1: *3304 to 8962GenBankGraphicsNext MatchPrevious Match
Alignment statistics for match #1
Score Expect Identities Gaps Strand
10394 bits(5628) 0.0 5650/5659(99%) 8/5659(0%) Plus/Plus
Range 2: *1859 to 2978GenBankGraphicsNext MatchPrevious MatchFirst Match
Alignment statistics for match #2
Score Expect Identities Gaps Strand
2036 bits(1102) 0.0 1115/1121(99%) 1/1121(0%) Plus/Plus
Range 4: *9281 to 9517GenBankGraphicsNext MatchPrevious MatchFirst Match
Alignment statistics for match #4
Score Expect Identities Gaps Strand
263 bits(142) 9e-64 209/241(87%) 6/241(2%) Plus/Plus
Y3 genome composition:
├─ Chromosome (~3.9 Mb): ✅ NO p2 integration detected
│ ├─ mash screen: empty (no genome-wide signal)
│ └─ blastn: single 44 bp hit = background/conserved motif
│
├─ Plasmid "3" (7,655 bp): 🟢 Identical to p1ATCC19606 (CP045108.1)
│
└─ p2-like plasmid: ❌ Completely absent (neither free nor integrated)
Biological implication:
Y3 carries ONLY the p1 lineage plasmid.
The p2 lineage is entirely missing from this isolate.
Only Y3 missing p2, report the detailed list of genes in the two plasmids, say Y3 is possibly missing p2, causing some phenotypical change, but tell my co-author should be very careful. Ask if the Y3 possibley missing p2 make sence? Write a comprehensive reports about the plasmid presence and absence?
-
Corrected Workflow: Systematic of two plasmids p1/p2 (p1ATCC19606 & p2ATCC19606) Screening for All 8 Samples