Protected: SanDisk High Endurance
Enter your password to view comments.
This post is a standalone, reproducible record of the bacterial WGS pipeline I used (example sample: AN6). I’m keeping all command lines (as-run) so you can reuse the workflow for future projects. Wherever you see absolute paths, replace them with your own.
-profile docker)fastqc, spades.py, shovill, pigz, awk, seqkit, fastANI, plus R (for plotting), and the tools required by the provided scripts. # Download the kmerfinder database: https://www.genomicepidemiology.org/services/ --> https://cge.food.dtu.dk/services/KmerFinder/ --> https://cge.food.dtu.dk/services/KmerFinder/etc/kmerfinder_db.tar.gz
# Download 20190108_kmerfinder_stable_dirs.tar.gz from https://zenodo.org/records/13447056
#--kmerfinderdb /path/to/kmerfinder/bacteria.tar.gz
#--kmerfinderdb /mnt/nvme1n1p1/REFs/kmerfinder_db.tar.gz
#--kmerfinderdb /mnt/nvme1n1p1/REFs/20190108_kmerfinder_stable_dirs.tar.gz
nextflow run nf-core/bacass -r 2.5.0 -profile docker \
--input samplesheet.tsv \
--outdir bacass_out \
--assembly_type long \
--kraken2db /mnt/nvme1n1p1/REFs/k2_standard_08_GB_20251015.tar.gz \
--kmerfinderdb /mnt/nvme1n1p1/REFs/kmerfinder/bacteria/ \
-resume
#SAVE bacass_out/Kmerfinder/kmerfinder_summary.csv to bacass_out/Kmerfinder/An6/An6_kmerfinder_results.xlsxln -s ../X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz An6_R1.fastq.gz
ln -s ../X101SC25116512-Z01-J002/01.RawData/An6/An6_2.fq.gz An6_R2.fastq.gz
mkdir fastqc_out
fastqc -t 4 raw_data/* -o fastqc_out/
mamba activate /home/jhuang/miniconda3/envs/bengal3_ac3For the AN6 data, it’s not better to run Trimmomatic first in most cases (adapters OK; per-tile failures are instrument/tile related and not “fixed” by trimming).
* **Adapters:** FastQC shows **Adapter Content = PASS** for both R1/R2. * **Overrepresented sequences:** none detected. * **Per-tile sequence quality:** **FAIL** (this is usually an instrument/tile effect; trimming adapters won’t “fix” it).Shovill: avoid pre-trimming (default read trimming already included unless disabled). SPAdes: trimming optional; try raw first, then trimmed if needed.
# Paired-end trimming with Trimmomatic (Illumina-style)
# Adjust TRIMMOMATIC_JAR and ADAPTERS paths to your install.
TRIMMOMATIC_JAR=/path/to/trimmomatic.jar
ADAPTERS=/path/to/Trimmomatic/adapters/TruSeq3-PE.fa
java -jar "$TRIMMOMATIC_JAR" PE -threads 16 -phred33 \
An6_R1.fastq.gz An6_R2.fastq.gz \
An6_R1.trim.paired.fastq.gz An6_R1.trim.unpaired.fastq.gz \
An6_R2.trim.paired.fastq.gz An6_R2.trim.unpaired.fastq.gz \
ILLUMINACLIP:"$ADAPTERS":2:30:10 \
LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:20 \
MINLEN:50What you feed into SPAdes/Shovill afterward:
Use the paired outputs:
An6_R1.trim.paired.fastq.gzAn6_R2.trim.paired.fastq.gzIf you want, I can also give the matching SPAdes command that includes unpaired reads (or the cleanest approach that ignores them).
spades.py \
-1 raw_data/An6_R1.fastq.gz \
-2 raw_data/An6_R2.fastq.gz \
--isolate \
-t 32 -m 250 \
-o spades_out
spades.py \
-1 raw_data/An6_R1.fastq.gz \
-2 raw_data/An6_R2.fastq.gz \
--careful \
-t 32 -m 250 \
-o spades_out_carefulShovill (CHOSEN; default does read trimming unless you disable it):
shovill \
--R1 raw_data/An6_R1.fastq.gz \
--R2 raw_data/An6_R2.fastq.gz \
--outdir shovill_out \
--cpus 32 --ram 250 \
--depth 100If you want to keep reads completely untrimmed in Shovill, add --noreadtrim.
# 1. Go up one level to the main 'bacass_out' directory
cd ..
# 2. Create directories for CheckM inputs and outputs
mkdir -p checkm_input checkm_output
# 3. Copy all .fna files into the 'checkm_input' folder
# (CheckM cannot search subdirectories, so they must be in one folder)
find ./Prokka -name "*.fna" -exec cp {} checkm_input/ \;
# 4. Run CheckM on all 4 assemblies
(checkm_env2) checkm lineage_wf -x fna checkm_input checkm_output
#Finished parsing hits for 4 of 4 (100.00%) bins.
#------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
#Bin Id Marker lineage # genomes # markers # marker sets 0 1 2 3 4 5+ Completeness Contamination Strain heterogeneity
#------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
#adeIJ_chr_plasmids f__Moraxellaceae (UID4680) 86 689 365 2 687 0 0 0 0 99.63 0.00 0.00
#adeAB_chr_plasmids f__Moraxellaceae (UID4680) 86 689 365 2 687 0 0 0 0 99.63 0.00 0.00
#A6WT_chr_plasmids f__Moraxellaceae (UID4680) 86 689 365 2 687 0 0 0 0 99.63 0.00 0.00
#A10CraA_chr_plasmids f__Moraxellaceae (UID4680) 86 689 365 2 687 0 0 0 0 99.63 0.00 0.00
#------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ # 1. 创建环境(推荐 mamba)
mamba create -n gtdbtk -c conda-forge -c bioconda gtdbtk
mamba activate gtdbtk
# 2. 下载数据库(仅需首次,约 60GB)
gtdbtk download --data_dir ./gtdb_data --release 220
wget https://data.gtdb.aau.ecogenomic.org/releases/release232/232.0/auxillary_files/gtdbtk_package/full_package/gtdbtk_r232_data.tar.g
mamba env config vars set GTDBTK_DATA_PATH="/mnt/nvme4n1p1/gtdb_data/release232"
# 先退出当前环境,再重新激活
mamba deactivate
mamba activate gtdbtk
# 验证环境变量是否加载成功
echo $GTDBTK_DATA_PATH
# 应输出:/mnt/nvme4n1p1/gtdb_data/release232
# 3. 运行分类(你提供的命令 + 实用参数)
gtdbtk classify_wf \
--genome_dir ./checkm_input \
--out_dir gtdb_out \
--cpus 64 \
--extension .fna \
--prefix mygenome
# 4. 查看结果
cat gtdb_out/classify/mygenome.bac120.summary.tsv # 细菌结果* Use: https://www.bv-brc.org/app/Annotation
* Input: scaffolded results from bacass
* Output: ./GenomeReport.html (Overview table) + ./FullGenomeReport.html (Genome Assembly + Genome Annotation + Subsystem Analysis + Specialty Genes + Antimicrobial Resistance Genes + Phylogenetic Analysis); It is more comprehensive than ComprehensiveGenomeAnalysis generating ./FullGenomeReport.html.# Prepare environment and run the Table 1 (Summary of sequence data and genome features (env: gunc_env)) pipeline:
# activate the env that has openpyxl
mamba activate gunc_env
mamba install -n gunc_env -c conda-forge openpyxl -y
mamba deactivate
# STEP_1
ENV_NAME=gunc_env \
SAMPLE=AN6 \
ASM=shovill_out/contigs.fa \
R1=./X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz \
R2=./X101SC25116512-Z01-J002/01.RawData/An6/An6_2.fq.gz \
~/Scripts/make_table1_pe.sh
ENV_NAME=gunc_env \
SAMPLE=wildtype \
ASM=bacass_out/checkm_input/A6WT_chr_plasmids.fna \
R1=./raw_data/19606adeAB_R1.fastq.gz \
R2=./raw_data/19606adeAB_R2.fastq.gz \
~/Scripts/make_table1_pe.sh
# A6WT_chr_plasmids.fna is scaffolded sequence and contains only two contigs, should use A6WT_contigs.min500.fasta
ENV_NAME=gunc_env \
SAMPLE=wildtype \
ASM=shovill/A6WT_contigs.min500.fasta \
R1=./raw_data/A6WT_R1.fastq.gz \
R2=./raw_data/A6WT_R2.fastq.gz \
~/Scripts/make_table1_pe.sh
# STEP_2
python export_table1_stats_to_excel_py36_compat.py \
--workdir table1_AN6_work \
--out Comprehensive_AN6.xlsx \
--max-rows 200000 \
--sample AN6
python ~/Scripts/export_table1_stats_to_excel_py36_compat.py \
--workdir table1_wildtype_work \
--out Comprehensive_wildtype.xlsx \
--max-rows 200000 \
--sample wildtype# -- The read number of raw data is not exact, using properly paired better! --
#zcat ./raw_data/A6WT_R1.fastq.gz | echo $(( $(wc -l) / 4 )) #--> 4,105,859 × 2
#pigz -dc X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz | awk 'END{print NR/4}'
#seqkit stats X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz
#seqkit stats ./raw_data/A6WT_R1.fastq.gz
##file format type num_seqs sum_len min_len avg_len max_len
##./raw_data/A6WT_R1.fastq.gz FASTQ DNA 4,105,859 619,984,709 151 151 151
# -- More advanced valid total number of reads sequenced using properly paired --
# 查看每个样本的比对率(看有多少reads成功比对)
samtools flagstat ./snippy/A6WT/A6WT.bam
samtools flagstat ./snippy/19606adeAB/19606adeAB.bam
samtools flagstat ./snippy/adeIJ/adeIJ.bam
samtools flagstat ./snippy/A10CraA/A10CraA.bam
6885728 + 0 properly paired (96.65% : N/A)
18895770 + 0 properly paired (89.77% : N/A)
17178378 + 0 properly paired (83.31% : N/A)
3577238 + 0 properly paired (58.36% : N/A)
# 查看参考基因组的大小(看是不是基因组本身不一样大)
samtools view -H ./snippy/A6WT/A6WT.bam | grep '^@SQ' | awk '{sum+=$3} END {print sum}'
samtools view -H ./snippy/A10CraA/A10CraA.bam | grep '^@SQ' | awk '{sum+=$3} END {print sum}'
# -- Calculate exact coverage from aligned BAM files (Recommended for manuscript) --
python3 ~/Scripts/calculate_coverage.py ./table1_wildtype_work/map/wildtype.bam
python3 ~/Scripts/calculate_coverage.py ./table1_adeAB_work/map/adeAB.bam
python3 ~/Scripts/calculate_coverage.py ./table1_adeIJ_work/map/adeIJ.bam
python3 ~/Scripts/calculate_coverage.py ./table1_craA_work/map/craA.bam
Average coverage: 310.99
Average coverage: 955.33
Average coverage: 886.04
Average coverage: 79.70
python3 ~/Scripts/calculate_coverage.py ./snippy/A6WT/A6WT.bam
python3 ~/Scripts/calculate_coverage.py ./snippy/19606adeAB/19606adeAB.bam
python3 ~/Scripts/calculate_coverage.py ./snippy/adeIJ/adeIJ.bam
python3 ~/Scripts/calculate_coverage.py ./snippy/A10CraA/A10CraA.bam
Average coverage: 262.93
Average coverage: 720.27
Average coverage: 653.39
Average coverage: 136.70| 样本 | Properly paired reads | Properly paired pairs(÷2) |
|---|---|---|
| A6WT | 6,885,728 | 3,442,864 |
| adeAB | 18,895,770 | 9,447,885 |
| adeIJ | 17,178,378 | 8,589,189 |
| A10CraA | 3,577,238 | 1,788,619 |
| 样本 | Properly paired pairs | 覆盖度 | 每个pair贡献的覆盖度 |
|---|---|---|---|
| A6WT | 3,442,864 | 262.93× | 262.93 / 3,442,864 ≈ 0.000076× per pair |
| adeAB | 9,447,885 | 720.27× | 720.27 / 9,447,885 ≈ 0.000076× per pair |
| adeIJ | 8,589,189 | 653.39× | 653.39 / 8,589,189 ≈ 0.000076× per pair |
| A10CraA | 1,788,619 | 136.70× | 136.70 / 1,788,619 ≈ 0.000076× per pair |
看到没有?每个pair贡献的覆盖度完全一致(都是 ~0.000076×)!
这进一步证明了:
A6WT: 3,442,864 pairs → 262.93×
A10CraA: 1,788,619 pairs → 136.70×
比值: 1,788,619 / 3,442,864 = 0.5195 ≈ 52%
比值: 136.70 / 262.93 = 0.5198 ≈ 52%完美对应! 😊
kate ./Manuscript2_MRA_Yan_RNA_chloramphenicol/A6WT_chr_plasmids.bgpipe.output_2799988.gb
COMMENT The annotation was added by the NCBI Prokaryotic Genome Annotation
Pipeline (PGAP). Information about PGAP can be found here:
https://www.ncbi.nlm.nih.gov/genome/annotation_prok/
##Genome-Assembly-Data-START##
Assembly Method :: SPAdes v. v3.15.5
Genome Representation :: Full
Expected Final Version :: No
Genome Coverage :: 360x
Sequencing Technology :: Illumina
##Genome-Assembly-Data-END##
##Genome-Annotation-Data-START##
Annotation Provider :: NCBI
Annotation Date :: 04/02/2026 16:34:58
Annotation Pipeline :: NCBI Prokaryotic Genome
Annotation Pipeline (PGAP)
Annotation Method :: Best-placed reference protein
set; GeneMarkS-2+
Annotation Software revision :: 6.10
Features Annotated :: Gene; CDS; rRNA; tRNA; ncRNA
Genes (total) :: 3,731 *
CDSs (total) :: 3,662
Genes (coding) :: 3,609
CDSs (with protein) :: 3,609 *
Genes (RNA) :: 69
rRNAs :: 1, 1, 1 (5S, 16S, 23S) *
complete rRNAs :: 1, 1, 1 (5S, 16S, 23S)
tRNAs :: 62 *
ncRNAs :: 4
Pseudo Genes (total) :: 53
CDSs (without protein) :: 53
Pseudo Genes (ambiguous residues) :: 0 of 53
Pseudo Genes (frameshifted) :: 24 of 53
Pseudo Genes (incomplete) :: 23 of 53
Pseudo Genes (internal stop) :: 11 of 53
Pseudo Genes (multiple problems) :: 5 of 53
CRISPR Arrays :: 1
##Genome-Annotation-Data-END##| Feature | GUNC (Used in your script) | EvalCon (BV-BRC Platform) |
|---|---|---|
| Full Name | Genome UNCluttered | Evaluation of Consistency (BV-BRC internal tool) |
| What it measures | Taxonomic / Phylogenetic Consistency | Functional / Metabolic Consistency |
| Core Algorithm | Maps genes to a reference phylogenetic tree (GTDB/proGenomes). Checks if genes in the genome come from a single evolutionary lineage or are a mix (chimeras/HGT). | Uses a machine-learning-derived catalog of ~1,300 functional roles (enzymes/pathways). Checks if the genome has biologically “expected” combinations of functions. |
| Coarse vs. Fine | Coarse: High taxonomic ranks (Kingdom, Phylum, Class). Fine: Low taxonomic ranks (Order, Family, Genus, Species). |
Evaluates consistency across different functional categories and pathway modules, not taxonomic ranks. |
| Primary Use Case | Detecting chimerism (e.g., in metagenome-assembled genomes, MAGs) and horizontal gene transfer. | Detecting functional anomalies or misannotations (e.g., a genome missing essential parts of a pathway it partially has). |
Conclusion for your paper: If you are writing a manuscript, you cannot say you used EvalCon if you actually ran GUNC. They measure different things. If your paper requires BV-BRC’s EvalG/EvalCon, you must run the BV-BRC pipeline.
Unlike CheckM or QUAST, EvalG and EvalCon are not standalone, easily downloadable command-line tools for local HPC clusters. They are proprietary/internal algorithms deeply integrated into the BV-BRC (Bacterial and Viral Bioinformatics Resource Center) annotation pipeline (which is based on the RAST annotation system).
To calculate them, you must use the BV-BRC ecosystem.
| Metric | Your Value | Tool | What it Measures | Interpretation of Your Example |
|---|---|---|---|---|
| Completeness | 100% | EvalG | The percentage of expected, lineage-specific single-copy marker genes present in the genome assembly. | Perfect. The genome contains 100% of the core structural marker genes expected for its taxonomic lineage, indicating a fully complete assembly. |
| Contamination | 0.2% | EvalG | The percentage of single-copy marker genes that appear in multiple copies, which suggests foreign DNA or a mixed assembly. | Excellent. Only 0.2% of markers are duplicated. The genome is virtually pure and free of contamination from other organisms. |
| Coarse Consistency | 99.5% | EvalCon | Functional coherence at a broad level. It checks if the genome possesses the expected major metabolic pathways and core biological subsystems for its taxonomy. | Excellent. 99.5% of the broad functional categories are logically consistent. No major biological pathways are inexplicably missing or broken. |
| Fine Consistency | 98.7% | EvalCon | Functional coherence at a granular level. It evaluates the predictable relationships between specific, individual enzymes and narrow functional roles (using a catalog of ~1,300 roles). | Very Good. 98.7% of the fine-grained functional roles align with expected biological relationships. The slight drop from coarse to fine is normal and represents minor, highly specific annotation gaps. |
If you are writing a paper, these four values together indicate that you have a near-perfect, high-quality genome assembly and annotation.
Example sentence for your Methods/Results section:
“Genome quality was assessed using the BV-BRC platform. EvalG confirmed a highly complete (100%) and pure (0.2% contamination) assembly based on lineage-specific single-copy markers. Furthermore, EvalCon demonstrated excellent functional coherence, with coarse and fine consistency scores of 99.5% and 98.7%, respectively, indicating that the annotated functional roles are highly consistent with expected metabolic pathways.”
Metricsa Value
Genome size (bp) 3,012,410
Contig count (>= 500 bp) 41
Total number of reads sequenced 15,929,405 × 2
Coverage depth (sequencing depth) 1454.3×
Coarse consistency (%) 99.67
Fine consistency (%) 94.50
Completeness (%) 99.73
Contamination (%) 0.21
Contigs N50 (bp) 169,757
Contigs L50 4
Guanine-cytosine content (%) 41.14
Number of coding sequences (CDSs) 2,938
Number of tRNAs 69
Number of rRNAs 3 cp shovill_out/contigs.fa AN6.fasta
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ASM=AN6.fasta SAMPLE=AN6 THREADS=32 ./run_resistome_virulome_dedup.sh #Default MINID=90 MINCOV=60
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ASM=AN6.fasta SAMPLE=AN6 MINID=80 MINCOV=60 ./run_resistome_virulome_dedup.sh # 0 0 0 0
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ASM=AN6.fasta SAMPLE=AN6 MINID=70 MINCOV=50 ./run_resistome_virulome_dedup.sh # 5 5 0 4
#Sanity checks on ABRicate outputs
grep -vc '^#' resistome_virulence_AN6/raw/AN6.megares.tab
grep -vc '^#' resistome_virulence_AN6/raw/AN6.card.tab
grep -vc '^#' resistome_virulence_AN6/raw/AN6.resfinder.tab
grep -vc '^#' resistome_virulence_AN6/raw/AN6.vfdb.tab
#!!!!!! DEBUG_TOMORROW: why using 'MINID=70 MINCOV=50' didn't return the 5504?
#Dedup tables / “one per gene” mode
rm Resistome_Virulence_An6.xlsx
chmod +x run_abricate_resistome_virulome_one_per_gene.sh
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 \
ASM=AN6.fasta \
SAMPLE=AN6 \
OUTDIR=resistome_virulence_AN6 \
MINID=70 MINCOV=50 \
THREADS=32 \
~/Scripts/run_abricate_resistome_virulome_one_per_gene.sh
cd resistome_virulence_AN6
python3 -c 'import pandas as pd; from pathlib import Path; files=["Table_AMR_genes_dedup.tsv","Table_AMR_genes_one_per_gene.tsv","Table_Virulence_VFDB_dedup.tsv","Table_DB_hit_counts.tsv"]; out="AN6_resistome_virulence.xlsx"; w=pd.ExcelWriter(out, engine="openpyxl"); [pd.read_csv(f, sep="\t").to_excel(w, sheet_name=Path(f).stem[:31], index=False) for f in files]; w.close(); print(out)' #Generate targets.tsv from ./bvbrc_out/Acinetobacter_harbinensis_AN6/FullGenomeReport.html.
export NCBI_EMAIL="xxx@yyy.de"
./resolve_best_assemblies_entrez.py targets.tsv resolved_accessions.tsv
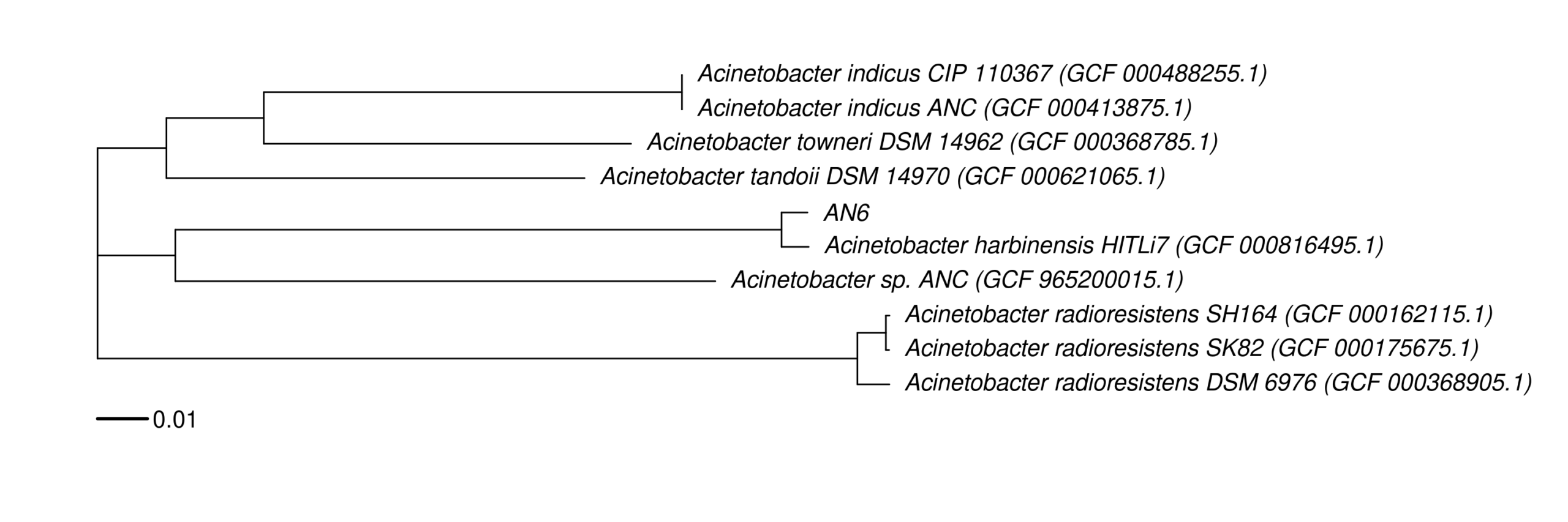
#[OK] Acinetobacter_harbinensis_HITLi7 -> GCF_000816495.1 (Scaffold)
#[OK] Acinetobacter_sp._ANC -> GCF_965200015.1 (Complete Genome)
#[OK] Acinetobacter_sp._TTH0-4 -> GCF_965200015.1 (Complete Genome)
#[OK] Acinetobacter_tandoii_DSM_14970 -> GCF_000621065.1 (Scaffold)
#[OK] Acinetobacter_towneri_DSM_14962 -> GCF_000368785.1 (Scaffold)
#[OK] Acinetobacter_radioresistens_SH164 -> GCF_000162115.1 (Scaffold)
#[OK] Acinetobacter_radioresistens_SK82 -> GCF_000175675.1 (Contig)
#[OK] Acinetobacter_radioresistens_DSM_6976 -> GCF_000368905.1 (Scaffold)
#[OK] Acinetobacter_indicus_ANC -> GCF_000413875.1 (Scaffold)
#[OK] Acinetobacter_indicus_CIP_110367 -> GCF_000488255.1 (Scaffold)
#NOTE the env bengal3_ac3 don’t have the following R package, using r_env for the plot-step → RUN TWICE, first bengal3_ac3, then run build_wgs_tree_fig3B.sh plot-only.
#ADAPT the params EXTRA_ASSEMBLIES (could stay as empty), and AN6.fasta as REF_FASTA
conda activate /home/jhuang/miniconda3/envs/bengal3_ac3
export NCBI_EMAIL="xxx@yyy.de"
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ./build_wgs_tree_fig3B.sh
# (Optional) if want to delete some leaves from the tree, remove from inputs so Roary cannot include it
for id in "GCF_002291425.1" "GCF_047901425.1" "GCF_004342245.1" "GCA_032062225.1"; do
rm -f work_wgs_tree/gffs/${id}.gff
rm -f work_wgs_tree/fastas/${id}.fna
rm -rf work_wgs_tree/prokka/${id}
rm -rf work_wgs_tree/genomes_ncbi/${id}
# remove from accession list so it won't come back
awk -F'\t' 'NR==1 || $2!="${id}"' work_wgs_tree/meta/accessions.tsv > work_wgs_tree/meta/accessions.tsv.tmp \
&& mv work_wgs_tree/meta/accessions.tsv.tmp work_wgs_tree/meta/accessions.tsv
done
./build_wgs_tree_fig3B.sh
#Wrote: work_wgs_tree/plot/labels.tsv
#Error: package or namespace load failed for ‘ggtree’ in loadNamespace(j <- i[[1L]], c(lib.loc, .libPaths()), versionCheck = vI[[j]]):
#there is no package called ‘aplot’
#Execution halted --> Using env r_env instead (see below)!
# Run this to regenerate labels.tsv
bash regenerate_labels.sh
# Regenerate the plot --> ERROR --> Using Rscript instead (see below)!
ENV_NAME=/home/jhuang/mambaforge/envs/r_env ./build_wgs_tree_fig3B.sh plot-only
#-->Error in as.hclust.phylo(tr) : the tree is not ultrametric
# 8) Manual correct the display name in work_wgs_tree/plot/labels.tsv
#sample display
#GCF_000816495.1 Acinetobacter harbinensis HITLi7 (GCF_000816495.1)
#GCF_965200015.1 Acinetobacter sp. ANC (GCF_965200015.1)
#GCF_000621065.1 Acinetobacter tandoii DSM 14970 (GCF_000621065.1)
#GCF_000368785.1 Acinetobacter towneri DSM 14962 (GCF_000368785.1)
#GCF_000162115.1 Acinetobacter radioresistens SH164 (GCF_000162115.1)
#GCF_000175675.1 Acinetobacter radioresistens SK82 (GCF_000175675.1)
#GCF_000368905.1 Acinetobacter radioresistens DSM 6976 (GCF_000368905.1)
#GCF_000413875.1 Acinetobacter indicus ANC (GCF_000413875.1)
#GCF_000488255.1 Acinetobacter indicus CIP 110367 (GCF_000488255.1)
#REF AN6
# 9) Rerun only the plot step uisng plot_tree_v4.R
Rscript ./plot_tree_v4.R \
work_wgs_tree/raxmlng/core.raxml.support \
work_wgs_tree/plot/labels.tsv \
6 \
work_wgs_tree/plot/core_tree.pdf \
work_wgs_tree/plot/core_tree.png mamba activate /home/jhuang/miniconda3/envs/bengal3_ac3
for id in GCF_000621065.1.fna GCF_000368785.1.fna GCF_000175675.1.fna GCF_000368905.1.fna GCF_000816495.1.fna GCF_965200015.1.fna GCF_000488255.1.fna GCF_000413875.1.fna GCF_000162115.1.fna; do
fastANI -q AN6.fasta -r ./work_wgs_tree/fastas/${id} -o fastANI_AN6_vs_${id}.txt
done
# Alternatively, we can use the script run_fastani_batch_verbose.sh.In total, we obtained 41 contigs >500 nt. Of these, 36 contigs were scaffolded with Multi-CSAR v1.1 into three chromosomal scaffolds:
The remaining five contigs (contig00026/32/33/37/39) could not be scaffolded. Their partial BLASTn matches to both plasmid and chromosomal sequences suggest shared mobile elements, but do not confirm circular plasmids. A sequence/assembly summary was exported to Excel (Summary_AN6.xlsx), including read yield/read-length statistics and key assembly/QC metrics (genome size, contigs/scaffolds, N50, GC%, completeness, contamination).
Below are the full scripts exactly as provided, including plot_tree_v4.R.
make_table1_pe.sh#!/usr/bin/env bash
set -Eeuo pipefail
# =========================
# User config
ENV_NAME="${ENV_NAME:-checkm_env2}"
# If you have Illumina paired-end, set R1/R2 (recommended)
R1="${R1:-}"
R2="${R2:-}"
# If you have single-end/ONT-like reads, set READS instead (legacy mode)
READS="${READS:-}"
ASM="${ASM:-shovill_out/contigs.fa}"
SAMPLE="${SAMPLE:-An6}"
THREADS="${THREADS:-32}"
OUT_TSV="${OUT_TSV:-Table1_${SAMPLE}.tsv}"
WORKDIR="${WORKDIR:-table1_${SAMPLE}_work}"
LOGDIR="${LOGDIR:-${WORKDIR}/logs}"
LOGFILE="${LOGFILE:-${LOGDIR}/run_$(date +%F_%H%M%S).log}"
AUTO_INSTALL="${AUTO_INSTALL:-1}" # 1=install missing tools in ENV_NAME
GUNC_DB_KIND="${GUNC_DB_KIND:-progenomes}" # progenomes or gtdb
# =========================
mkdir -p "${LOGDIR}"
exec > >(tee -a "${LOGFILE}") 2>&1
ts(){ date +"%F %T"; }
log(){ echo "[$(ts)] $*"; }
on_err() {
local ec=$?
log "ERROR: failed (exit=${ec}) at line ${BASH_LINENO[0]}: ${BASH_COMMAND}"
log "Logfile: ${LOGFILE}"
exit "${ec}"
}
trap on_err ERR
# print every command
set -x
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
pick_pm() {
if need_cmd mamba; then echo "mamba"
elif need_cmd conda; then echo "conda"
else
log "ERROR: neither mamba nor conda found in PATH"
exit 1
fi
}
activate_env() {
if ! need_cmd conda; then
log "ERROR: conda not found; cannot activate env"
exit 1
fi
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
conda activate "${ENV_NAME}"
}
ensure_env_exists() {
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
if ! conda env list | awk '{print $1}' | grep -qx "${ENV_NAME}"; then
log "ERROR: env ${ENV_NAME} not found. Create it first."
exit 1
fi
}
install_pkgs_in_env() {
local pm="$1"; shift
local pkgs=("$@")
log "Installing into env ${ENV_NAME}: ${pkgs[*]}"
"${pm}" install -n "${ENV_NAME}" -c bioconda -c conda-forge -y "${pkgs[@]}"
}
pick_quast_cmd() {
if need_cmd quast; then echo "quast"
elif need_cmd quast.py; then echo "quast.py"
else echo ""
fi
}
# tool->package mapping (install missing ones)
declare -A TOOL2PKG=(
[quast]="quast"
[minimap2]="minimap2"
[samtools]="samtools"
[mosdepth]="mosdepth"
[checkm]="checkm-genome=1.1.3"
[gunc]="gunc"
[python]="python"
)
# =========================
# Detect mode (PE vs single)
MODE=""
if [[ -n "${R1}" || -n "${R2}" ]]; then
[[ -n "${R1}" && -n "${R2}" ]] || { log "ERROR: Provide both R1 and R2."; exit 1; }
MODE="PE"
elif [[ -n "${READS}" ]]; then
MODE="SINGLE"
else
log "ERROR: Provide either (R1+R2) OR READS."
exit 1
fi
# =========================
# Start
log "Start: Table 1 generation (reuse env=${ENV_NAME})"
log "Assembly: ${ASM}"
log "Sample: ${SAMPLE}"
log "Threads: ${THREADS}"
log "Workdir: ${WORKDIR}"
log "Logfile: ${LOGFILE}"
log "Mode: ${MODE}"
if [[ "${MODE}" == "PE" ]]; then
log "R1: ${R1}"
log "R2: ${R2}"
else
log "Reads: ${READS}"
fi
PM="$(pick_pm)"
log "Pkg manager: ${PM}"
ensure_env_exists
activate_env
log "Active envs:"
conda info --envs
log "Versions (if available):"
( python --version || true )
( checkm --version || true )
( gunc -v || true )
( minimap2 --version 2>&1 | head -n 2 || true )
( samtools --version 2>&1 | head -n 2 || true )
( mosdepth --version 2>&1 | head -n 2 || true )
( quast --version 2>&1 | head -n 2 || true )
( quast.py --version 2>&1 | head -n 2 || true )
# =========================
# Check/install missing tools in this env
MISSING_PKGS=()
for tool in minimap2 samtools mosdepth checkm gunc python; do
if ! need_cmd "${tool}"; then
MISSING_PKGS+=("${TOOL2PKG[$tool]}")
fi
done
QUAST_CMD="$(pick_quast_cmd)"
if [[ -z "${QUAST_CMD}" ]]; then
MISSING_PKGS+=("${TOOL2PKG[quast]}")
fi
if [[ "${#MISSING_PKGS[@]}" -gt 0 ]]; then
if [[ "${AUTO_INSTALL}" != "1" ]]; then
log "ERROR: missing tools and AUTO_INSTALL=0. Missing packages: ${MISSING_PKGS[*]}"
exit 1
fi
mapfile -t UNIQUE < <(printf "%s\n" "${MISSING_PKGS[@]}" | awk '!seen[$0]++')
install_pkgs_in_env "${PM}" "${UNIQUE[@]}"
activate_env
QUAST_CMD="$(pick_quast_cmd)"
fi
for tool in minimap2 samtools mosdepth checkm gunc python; do
need_cmd "${tool}" || { log "ERROR: still missing tool: ${tool}"; exit 1; }
done
[[ -n "${QUAST_CMD}" ]] || { log "ERROR: QUAST still missing."; exit 1; }
log "All tools ready. QUAST cmd: ${QUAST_CMD}"
# =========================
# Prepare workdir
mkdir -p "${WORKDIR}"/{genomes,reads,stats,quast,map,checkm,gunc,tmp}
ASM_ABS="$(realpath "${ASM}")"
ln -sf "${ASM_ABS}" "${WORKDIR}/genomes/${SAMPLE}.fasta"
if [[ "${MODE}" == "PE" ]]; then
R1_ABS="$(realpath "${R1}")"
R2_ABS="$(realpath "${R2}")"
ln -sf "${R1_ABS}" "${WORKDIR}/reads/${SAMPLE}.R1.fastq.gz"
ln -sf "${R2_ABS}" "${WORKDIR}/reads/${SAMPLE}.R2.fastq.gz"
else
READS_ABS="$(realpath "${READS}")"
ln -sf "${READS_ABS}" "${WORKDIR}/reads/${SAMPLE}.reads.fastq.gz"
fi
# =========================
# 1) QUAST
log "Run QUAST..."
"${QUAST_CMD}" "${WORKDIR}/genomes/${SAMPLE}.fasta" -o "${WORKDIR}/quast"
QUAST_TSV="${WORKDIR}/quast/report.tsv"
test -s "${QUAST_TSV}"
# =========================
# 2) Map reads + mosdepth
log "Map reads (minimap2) + sort BAM..."
SORT_T="$((THREADS>16?16:THREADS))"
if [[ "${MODE}" == "PE" ]]; then
minimap2 -t "${THREADS}" -ax sr \
"${WORKDIR}/genomes/${SAMPLE}.fasta" \
"${WORKDIR}/reads/${SAMPLE}.R1.fastq.gz" "${WORKDIR}/reads/${SAMPLE}.R2.fastq.gz" \
| samtools sort -@ "${SORT_T}" -o "${WORKDIR}/map/${SAMPLE}.bam" -
else
# legacy single-read mode; keep map-ont as in original script
minimap2 -t "${THREADS}" -ax map-ont \
"${WORKDIR}/genomes/${SAMPLE}.fasta" "${WORKDIR}/reads/${SAMPLE}.reads.fastq.gz" \
| samtools sort -@ "${SORT_T}" -o "${WORKDIR}/map/${SAMPLE}.bam" -
fi
samtools index "${WORKDIR}/map/${SAMPLE}.bam"
log "Compute depth (mosdepth)..."
mosdepth -t "${SORT_T}" "${WORKDIR}/map/${SAMPLE}" "${WORKDIR}/map/${SAMPLE}.bam"
MOS_SUMMARY="${WORKDIR}/map/${SAMPLE}.mosdepth.summary.txt"
test -s "${MOS_SUMMARY}"
# =========================
# 3) CheckM
log "Run CheckM lineage_wf..."
checkm lineage_wf -x fasta -t "${THREADS}" "${WORKDIR}/genomes" "${WORKDIR}/checkm/out"
log "Run CheckM qa..."
checkm qa "${WORKDIR}/checkm/out/lineage.ms" "${WORKDIR}/checkm/out" --tab_table -o 2 \
> "${WORKDIR}/checkm/checkm_summary.tsv"
CHECKM_SUM="${WORKDIR}/checkm/checkm_summary.tsv"
test -s "${CHECKM_SUM}"
# =========================
# 4) GUNC
log "Run GUNC..."
mkdir -p "${WORKDIR}/gunc/db" "${WORKDIR}/gunc/out"
if [[ -z "$(ls -A "${WORKDIR}/gunc/db" 2>/dev/null || true)" ]]; then
log "Downloading GUNC DB kind=${GUNC_DB_KIND} to ${WORKDIR}/gunc/db ..."
gunc download_db -db "${GUNC_DB_KIND}" "${WORKDIR}/gunc/db"
fi
DMND="$(find "${WORKDIR}/gunc/db" -type f -name "*.dmnd" | head -n 1 || true)"
if [[ -z "${DMND}" ]]; then
log "ERROR: No *.dmnd found under ${WORKDIR}/gunc/db after download."
ls -lah "${WORKDIR}/gunc/db" || true
exit 1
fi
log "Using GUNC db_file: ${DMND}"
gunc run \
--db_file "${DMND}" \
--input_fasta "${WORKDIR}/genomes/${SAMPLE}.fasta" \
--out_dir "${WORKDIR}/gunc/out" \
--threads "${THREADS}" \
--detailed_output \
--contig_taxonomy_output \
--use_species_level
ALL_LEVELS="$(find "${WORKDIR}/gunc/out" -name "*all_levels.tsv" | head -n 1 || true)"
test -n "${ALL_LEVELS}"
log "Found GUNC all_levels.tsv: ${ALL_LEVELS}"
# =========================
# 5) Parse outputs and write Table 1 TSV
log "Parse outputs → ${OUT_TSV}"
export SAMPLE WORKDIR OUT_TSV GUNC_ALL_LEVELS="${ALL_LEVELS}"
python - <<'PY'
import csv, os
sample = os.environ["SAMPLE"]
workdir = os.environ["WORKDIR"]
out_tsv = os.environ["OUT_TSV"]
gunc_all_levels = os.environ["GUNC_ALL_LEVELS"]
quast_tsv = os.path.join(workdir, "quast", "report.tsv")
mos_summary = os.path.join(workdir, "map", f"{sample}.mosdepth.summary.txt")
checkm_sum = os.path.join(workdir, "checkm", "checkm_summary.tsv")
def read_quast(path):
with open(path, newline="") as f:
rows = list(csv.reader(f, delimiter="\t"))
asm_idx = 1
d = {}
for r in rows[1:]:
if not r: continue
key = r[0].strip()
val = r[asm_idx].strip() if asm_idx < len(r) else ""
d[key] = val
return d
def read_mosdepth(path):
with open(path) as f:
for line in f:
if line.startswith("chrom"): continue
parts = line.rstrip("\n").split("\t")
if len(parts) >= 4 and parts[0] == "total":
return parts[3]
return ""
def read_checkm(path, sample):
with open(path, newline="") as f:
reader = csv.DictReader(f, delimiter="\t")
for row in reader:
bid = row.get("Bin Id") or row.get("Bin") or row.get("bin_id") or ""
if bid == sample:
return row
return {}
def read_gunc_all_levels(path):
coarse_lvls = {"kingdom","phylum","class"}
fine_lvls = {"order","family","genus","species"}
coarse, fine = [], []
best_line = None
rank = {"kingdom":0,"phylum":1,"class":2,"order":3,"family":4,"genus":5,"species":6}
best_rank = -1
with open(path, newline="") as f:
reader = csv.DictReader(f, delimiter="\t")
for row in reader:
lvl = (row.get("taxonomic_level") or "").strip()
p = row.get("proportion_genes_retained_in_major_clades") or ""
try:
pv = float(p)
except:
pv = None
if pv is not None:
if lvl in coarse_lvls: coarse.append(pv)
if lvl in fine_lvls: fine.append(pv)
if lvl in rank and rank[lvl] > best_rank:
best_rank = rank[lvl]
best_line = row
coarse_mean = sum(coarse)/len(coarse) if coarse else ""
fine_mean = sum(fine)/len(fine) if fine else ""
contamination_portion = best_line.get("contamination_portion","") if best_line else ""
pass_gunc = best_line.get("pass.GUNC","") if best_line else ""
return coarse_mean, fine_mean, contamination_portion, pass_gunc
qu = read_quast(quast_tsv)
mean_depth = read_mosdepth(mos_summary)
ck = read_checkm(checkm_sum, sample)
coarse_mean, fine_mean, contamination_portion, pass_gunc = read_gunc_all_levels(gunc_all_levels)
header = [
"Sample",
"Genome_length_bp",
"Contigs",
"N50_bp",
"L50",
"GC_percent",
"Mean_depth_x",
"CheckM_completeness_percent",
"CheckM_contamination_percent",
"CheckM_strain_heterogeneity_percent",
"GUNC_coarse_consistency",
"GUNC_fine_consistency",
"GUNC_contamination_portion",
"GUNC_pass"
]
row = [
sample,
qu.get("Total length", ""),
qu.get("# contigs", ""),
qu.get("N50", ""),
qu.get("L50", ""),
qu.get("GC (%)", ""),
mean_depth,
ck.get("Completeness", ""),
ck.get("Contamination", ""),
ck.get("Strain heterogeneity", ""),
f"{coarse_mean:.4f}" if isinstance(coarse_mean, float) else coarse_mean,
f"{fine_mean:.4f}" if isinstance(fine_mean, float) else fine_mean,
contamination_portion,
pass_gunc
]
with open(out_tsv, "w", newline="") as f:
w = csv.writer(f, delimiter="\t")
w.writerow(header)
w.writerow(row)
print(f"OK: wrote {out_tsv}")
PY
log "SUCCESS"
log "Output TSV: ${OUT_TSV}"
log "Workdir: ${WORKDIR}"
log "Logfile: ${LOGFILE}"export_table1_stats_to_excel_py36_compat.py#!/usr/bin/env python3
# -*- coding: utf-8 -*-
"""
Export a comprehensive Excel workbook from a Table1 pipeline workdir.
Python 3.6 compatible (no PEP604 unions, no builtin generics).
Requires: openpyxl
Sheets (as available):
- Summary
- Table1 (if Table1_*.tsv exists)
- QUAST_report (report.tsv)
- QUAST_metrics (metric/value)
- Mosdepth_summary (*.mosdepth.summary.txt)
- CheckM (checkm_summary.tsv)
- GUNC_* (all .tsv under gunc/out)
- File_Inventory (relative path, size, mtime; optional md5 for small files)
- Run_log_preview (head/tail of latest log under workdir/logs or workdir/*/logs)
"""
from __future__ import print_function
import argparse
import csv
import hashlib
import os
import sys
import time
from pathlib import Path
try:
from openpyxl import Workbook
from openpyxl.utils import get_column_letter
except ImportError:
sys.stderr.write("ERROR: openpyxl is required. Install with:\n"
" conda install -c conda-forge openpyxl\n")
raise
MAX_XLSX_ROWS = 1048576
def safe_sheet_name(name, used):
# Excel: <=31 chars, cannot contain: : \ / ? * [ ]
bad = r'[:\\/?*\[\]]'
base = name.strip() or "Sheet"
base = __import__("re").sub(bad, "_", base)
base = base[:31]
if base not in used:
used.add(base)
return base
# make unique with suffix
for i in range(2, 1000):
suffix = "_%d" % i
cut = 31 - len(suffix)
candidate = (base[:cut] + suffix)
if candidate not in used:
used.add(candidate)
return candidate
raise RuntimeError("Too many duplicate sheet names for base=%s" % base)
def autosize(ws, max_width=60):
for col in ws.columns:
max_len = 0
col_letter = get_column_letter(col[0].column)
for cell in col:
v = cell.value
if v is None:
continue
s = str(v)
if len(s) > max_len:
max_len = len(s)
ws.column_dimensions[col_letter].width = min(max_width, max(10, max_len + 2))
def write_table(ws, header, rows, max_rows=None):
if header:
ws.append(header)
count = 0
for r in rows:
ws.append(r)
count += 1
if max_rows is not None and count >= max_rows:
break
def read_tsv(path, max_rows=None):
header = []
rows = []
with path.open("r", newline="") as f:
reader = csv.reader(f, delimiter="\t")
for i, r in enumerate(reader):
if i == 0:
header = r
continue
rows.append(r)
if max_rows is not None and len(rows) >= max_rows:
break
return header, rows
def read_text_table(path, max_rows=None):
# for mosdepth summary (tsv with header)
return read_tsv(path, max_rows=max_rows)
def md5_file(path, chunk=1024*1024):
h = hashlib.md5()
with path.open("rb") as f:
while True:
b = f.read(chunk)
if not b:
break
h.update(b)
return h.hexdigest()
def find_latest_log(workdir):
candidates = []
# common locations
for p in [workdir / "logs", workdir / "log", workdir / "Logs"]:
if p.exists():
candidates.extend(p.glob("*.log"))
# nested logs
candidates.extend(workdir.glob("**/logs/*.log"))
if not candidates:
return None
candidates.sort(key=lambda x: x.stat().st_mtime, reverse=True)
return candidates[0]
def add_summary_sheet(wb, used, info_items):
ws = wb.create_sheet(title=safe_sheet_name("Summary", used))
ws.append(["Key", "Value"])
for k, v in info_items:
ws.append([k, v])
autosize(ws)
def add_log_preview(wb, used, log_path, head_n=80, tail_n=120):
if log_path is None or not log_path.exists():
return
ws = wb.create_sheet(title=safe_sheet_name("Run_log_preview", used))
ws.append(["Log path", str(log_path)])
ws.append([])
lines = log_path.read_text(errors="replace").splitlines()
ws.append(["--- HEAD (%d) ---" % head_n])
for line in lines[:head_n]:
ws.append([line])
ws.append([])
ws.append(["--- TAIL (%d) ---" % tail_n])
for line in lines[-tail_n:]:
ws.append([line])
ws.column_dimensions["A"].width = 120
def add_file_inventory(wb, used, workdir, do_md5=True, md5_max_bytes=200*1024*1024, max_rows=None):
ws = wb.create_sheet(title=safe_sheet_name("File_Inventory", used))
ws.append(["relative_path", "size_bytes", "mtime_iso", "md5(optional)"])
count = 0
for p in sorted(workdir.rglob("*")):
if p.is_dir():
continue
rel = str(p.relative_to(workdir))
st = p.stat()
mtime = time.strftime("%Y-%m-%d %H:%M:%S", time.localtime(st.st_mtime))
md5 = ""
if do_md5 and st.st_size <= md5_max_bytes:
try:
md5 = md5_file(p)
except Exception:
md5 = "ERROR"
ws.append([rel, st.st_size, mtime, md5])
count += 1
if max_rows is not None and count >= max_rows:
break
autosize(ws, max_width=80)
def add_tsv_sheet(wb, used, name, path, max_rows=None):
header, rows = read_tsv(path, max_rows=max_rows)
ws = wb.create_sheet(title=safe_sheet_name(name, used))
write_table(ws, header, rows, max_rows=max_rows)
autosize(ws, max_width=80)
def add_quast_metrics_sheet(wb, used, quast_report_tsv):
header, rows = read_tsv(quast_report_tsv, max_rows=None)
if not header or len(header) < 2:
return
asm_name = header[1]
ws = wb.create_sheet(title=safe_sheet_name("QUAST_metrics", used))
ws.append(["Metric", asm_name])
for r in rows:
if not r:
continue
metric = r[0]
val = r[1] if len(r) > 1 else ""
ws.append([metric, val])
autosize(ws, max_width=80)
def main():
ap = argparse.ArgumentParser()
ap.add_argument("--workdir", required=True, help="workdir produced by pipeline (e.g., table1_GE11174_work)")
ap.add_argument("--out", required=True, help="output .xlsx")
ap.add_argument("--sample", default="", help="sample name for summary")
ap.add_argument("--max-rows", type=int, default=200000, help="max rows per large sheet")
ap.add_argument("--no-md5", action="store_true", help="skip md5 calculation in File_Inventory")
args = ap.parse_args()
workdir = Path(args.workdir).resolve()
out = Path(args.out).resolve()
if not workdir.exists():
sys.stderr.write("ERROR: workdir not found: %s\n" % workdir)
sys.exit(2)
wb = Workbook()
# remove default sheet
wb.remove(wb.active)
used = set()
# Summary info
info = [
("sample", args.sample or ""),
("workdir", str(workdir)),
("generated_at", time.strftime("%Y-%m-%d %H:%M:%S")),
("python", sys.version.replace("\n", " ")),
("openpyxl", __import__("openpyxl").__version__),
]
add_summary_sheet(wb, used, info)
# Table1 TSV (try common names)
table1_candidates = list(workdir.glob("Table1_*.tsv")) + list(workdir.glob("*.tsv"))
# Prefer Table1_*.tsv in workdir root
table1_path = None
for p in table1_candidates:
if p.name.startswith("Table1_") and p.suffix == ".tsv":
table1_path = p
break
if table1_path is None:
# maybe created in cwd, not inside workdir; try alongside workdir
parent = workdir.parent
for p in parent.glob("Table1_*.tsv"):
if args.sample and args.sample in p.name:
table1_path = p
break
if table1_path is None and list(parent.glob("Table1_*.tsv")):
table1_path = sorted(parent.glob("Table1_*.tsv"))[0]
if table1_path is not None and table1_path.exists():
add_tsv_sheet(wb, used, "Table1", table1_path, max_rows=args.max_rows)
# QUAST
quast_report = workdir / "quast" / "report.tsv"
if quast_report.exists():
add_tsv_sheet(wb, used, "QUAST_report", quast_report, max_rows=args.max_rows)
add_quast_metrics_sheet(wb, used, quast_report)
# Mosdepth summary
for p in sorted((workdir / "map").glob("*.mosdepth.summary.txt")):
# mosdepth summary is TSV-like
name = "Mosdepth_" + p.stem.replace(".mosdepth.summary", "")
add_tsv_sheet(wb, used, name[:31], p, max_rows=args.max_rows)
# CheckM
checkm_sum = workdir / "checkm" / "checkm_summary.tsv"
if checkm_sum.exists():
add_tsv_sheet(wb, used, "CheckM", checkm_sum, max_rows=args.max_rows)
# GUNC outputs (all TSV under gunc/out)
gunc_out = workdir / "gunc" / "out"
if gunc_out.exists():
for p in sorted(gunc_out.rglob("*.tsv")):
rel = str(p.relative_to(gunc_out))
sheet = "GUNC_" + rel.replace("/", "_").replace("\\", "_").replace(".tsv", "")
add_tsv_sheet(wb, used, sheet[:31], p, max_rows=args.max_rows)
# Log preview
latest_log = find_latest_log(workdir)
add_log_preview(wb, used, latest_log)
# File inventory
add_file_inventory(
wb, used, workdir,
do_md5=(not args.no_md5),
md5_max_bytes=200*1024*1024,
max_rows=args.max_rows
)
# Save
out.parent.mkdir(parents=True, exist_ok=True)
wb.save(str(out))
print("OK: wrote %s" % out)
if __name__ == "__main__":
main()run_resistome_virulome_dedup.sh#!/usr/bin/env bash
set -Eeuo pipefail
# -------- user inputs --------
ENV_NAME="${ENV_NAME:-bengal3_ac3}"
ASM="${ASM:-GE11174.fasta}"
SAMPLE="${SAMPLE:-GE11174}"
OUTDIR="${OUTDIR:-resistome_virulence_${SAMPLE}}"
THREADS="${THREADS:-16}"
# thresholds (set to 0/0 if you truly want ABRicate defaults)
MINID="${MINID:-90}"
MINCOV="${MINCOV:-60}"
# ----------------------------
log(){ echo "[$(date +'%F %T')] $*" >&2; }
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
activate_env() {
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
conda activate "${ENV_NAME}"
}
main(){
activate_env
mkdir -p "${OUTDIR}"/{raw,amr,virulence,card,tmp}
log "Env: ${ENV_NAME}"
log "ASM: ${ASM}"
log "Sample: ${SAMPLE}"
log "Outdir: ${OUTDIR}"
log "ABRicate thresholds: MINID=${MINID} MINCOV=${MINCOV}"
log "ABRicate DB list:"
abricate --list | egrep -i "vfdb|resfinder|megares|card" || true
# Make sure indices exist
log "Running abricate --setupdb (safe even if already done)..."
abricate --setupdb
# ---- ABRicate AMR DBs ----
log "Running ABRicate: ResFinder"
abricate --db resfinder --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
log "Running ABRicate: MEGARes"
abricate --db megares --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.megares.tab"
# ---- Virulence (VFDB) ----
log "Running ABRicate: VFDB"
abricate --db vfdb --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
# ---- CARD: prefer RGI if available, else ABRicate card ----
CARD_MODE="ABRicate"
if need_cmd rgi; then
log "RGI found. Trying RGI (CARD) ..."
set +e
rgi main --input_sequence "${ASM}" --output_file "${OUTDIR}/card/${SAMPLE}.rgi" --input_type contig --num_threads "${THREADS}"
rc=$?
set -e
if [[ $rc -eq 0 ]]; then
CARD_MODE="RGI"
else
log "RGI failed (likely CARD data not installed). Falling back to ABRicate card."
fi
fi
if [[ "${CARD_MODE}" == "ABRicate" ]]; then
log "Running ABRicate: CARD"
abricate --db card --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.card.tab"
fi
# ---- Build deduplicated tables ----
log "Creating deduplicated AMR/VFDB tables..."
export OUTDIR SAMPLE CARD_MODE
python - <<'PY'
import os, re
from pathlib import Path
import pandas as pd
from io import StringIO
outdir = Path(os.environ["OUTDIR"])
sample = os.environ["SAMPLE"]
card_mode = os.environ["CARD_MODE"]
def read_abricate_tab(path: Path, source: str) -> pd.DataFrame:
if not path.exists() or path.stat().st_size == 0:
return pd.DataFrame()
lines=[]
with path.open("r", errors="replace") as f:
for line in f:
if line.startswith("#") or not line.strip():
continue
lines.append(line)
if not lines:
return pd.DataFrame()
df = pd.read_csv(StringIO("".join(lines)), sep="\t", dtype=str)
df.insert(0, "Source", source)
return df
def to_num(s):
try:
return float(str(s).replace("%",""))
except:
return None
def normalize_abricate(df: pd.DataFrame, dbname: str) -> pd.DataFrame:
if df.empty:
return pd.DataFrame(columns=[
"Source","Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"
])
# Column names vary slightly; handle common ones
gene = "GENE" if "GENE" in df.columns else None
prod = "PRODUCT" if "PRODUCT" in df.columns else None
acc = "ACCESSION" if "ACCESSION" in df.columns else None
contig = "SEQUENCE" if "SEQUENCE" in df.columns else ("CONTIG" if "CONTIG" in df.columns else None)
start = "START" if "START" in df.columns else None
end = "END" if "END" in df.columns else None
strand= "STRAND" if "STRAND" in df.columns else None
pid = "%IDENTITY" if "%IDENTITY" in df.columns else ("% Identity" if "% Identity" in df.columns else None)
pcv = "%COVERAGE" if "%COVERAGE" in df.columns else ("% Coverage" if "% Coverage" in df.columns else None)
out = pd.DataFrame()
out["Source"] = df["Source"]
out["Database"] = dbname
out["Gene"] = df[gene] if gene else ""
out["Product"] = df[prod] if prod else ""
out["Accession"] = df[acc] if acc else ""
out["Contig"] = df[contig] if contig else ""
out["Start"] = df[start] if start else ""
out["End"] = df[end] if end else ""
out["Strand"] = df[strand] if strand else ""
out["Pct_Identity"] = df[pid] if pid else ""
out["Pct_Coverage"] = df[pcv] if pcv else ""
return out
def dedup_best(df: pd.DataFrame, key_cols):
"""Keep best hit per key by highest identity, then coverage, then longest span."""
if df.empty:
return df
# numeric helpers
df = df.copy()
df["_pid"] = df["Pct_Identity"].map(to_num)
df["_pcv"] = df["Pct_Coverage"].map(to_num)
def span(row):
try:
return abs(int(row["End"]) - int(row["Start"])) + 1
except:
return 0
df["_span"] = df.apply(span, axis=1)
# sort best-first
df = df.sort_values(by=["_pid","_pcv","_span"], ascending=[False,False,False], na_position="last")
df = df.drop_duplicates(subset=key_cols, keep="first")
df = df.drop(columns=["_pid","_pcv","_span"])
return df
# ---------- AMR inputs ----------
amr_frames = []
# ResFinder (often 0 hits; still okay)
resfinder = outdir / "raw" / f"{sample}.resfinder.tab"
df = read_abricate_tab(resfinder, "ABRicate")
amr_frames.append(normalize_abricate(df, "ResFinder"))
# MEGARes
megares = outdir / "raw" / f"{sample}.megares.tab"
df = read_abricate_tab(megares, "ABRicate")
amr_frames.append(normalize_abricate(df, "MEGARes"))
# CARD: RGI or ABRicate
if card_mode == "RGI":
# Try common RGI tab outputs
prefix = outdir / "card" / f"{sample}.rgi"
rgi_tab = None
for ext in [".txt",".tab",".tsv"]:
p = Path(str(prefix) + ext)
if p.exists() and p.stat().st_size > 0:
rgi_tab = p
break
if rgi_tab is not None:
rgi = pd.read_csv(rgi_tab, sep="\t", dtype=str)
out = pd.DataFrame()
out["Source"] = "RGI"
out["Database"] = "CARD"
# Prefer ARO_name/Best_Hit_ARO if present
out["Gene"] = rgi["ARO_name"] if "ARO_name" in rgi.columns else (rgi["Best_Hit_ARO"] if "Best_Hit_ARO" in rgi.columns else "")
out["Product"] = rgi["ARO_name"] if "ARO_name" in rgi.columns else ""
out["Accession"] = rgi["ARO_accession"] if "ARO_accession" in rgi.columns else ""
out["Contig"] = rgi["Sequence"] if "Sequence" in rgi.columns else ""
out["Start"] = rgi["Start"] if "Start" in rgi.columns else ""
out["End"] = rgi["Stop"] if "Stop" in rgi.columns else (rgi["End"] if "End" in rgi.columns else "")
out["Strand"] = rgi["Orientation"] if "Orientation" in rgi.columns else ""
out["Pct_Identity"] = rgi["% Identity"] if "% Identity" in rgi.columns else ""
out["Pct_Coverage"] = rgi["% Coverage"] if "% Coverage" in rgi.columns else ""
amr_frames.append(out)
else:
card = outdir / "raw" / f"{sample}.card.tab"
df = read_abricate_tab(card, "ABRicate")
amr_frames.append(normalize_abricate(df, "CARD"))
amr_all = pd.concat([x for x in amr_frames if not x.empty], ignore_index=True) if any(not x.empty for x in amr_frames) else pd.DataFrame(
columns=["Source","Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"]
)
# Deduplicate within each (Database,Gene) – this is usually what you want for manuscript tables
amr_dedup = dedup_best(amr_all, key_cols=["Database","Gene"])
# Sort nicely
if not amr_dedup.empty:
amr_dedup = amr_dedup.sort_values(["Database","Gene"]).reset_index(drop=True)
amr_out = outdir / "Table_AMR_genes_dedup.tsv"
amr_dedup.to_csv(amr_out, sep="\t", index=False)
# ---------- Virulence (VFDB) ----------
vfdb = outdir / "raw" / f"{sample}.vfdb.tab"
vf = read_abricate_tab(vfdb, "ABRicate")
vf_norm = normalize_abricate(vf, "VFDB")
# Dedup within (Gene) for VFDB (or use Database,Gene; Database constant)
vf_dedup = dedup_best(vf_norm, key_cols=["Gene"]) if not vf_norm.empty else vf_norm
if not vf_dedup.empty:
vf_dedup = vf_dedup.sort_values(["Gene"]).reset_index(drop=True)
vf_out = outdir / "Table_Virulence_VFDB_dedup.tsv"
vf_dedup.to_csv(vf_out, sep="\t", index=False)
print("OK wrote:")
print(" ", amr_out)
print(" ", vf_out)
PY
log "Done."
log "Outputs:"
log " ${OUTDIR}/Table_AMR_genes_dedup.tsv"
log " ${OUTDIR}/Table_Virulence_VFDB_dedup.tsv"
log "Raw:"
log " ${OUTDIR}/raw/${SAMPLE}.*.tab"
}
mainrun_abricate_resistome_virulome_one_per_gene.sh#!/usr/bin/env bash
set -Eeuo pipefail
# ------------------- USER SETTINGS -------------------
ENV_NAME="${ENV_NAME:-bengal3_ac3}"
ASM="${ASM:-GE11174.fasta}" # input assembly fasta
SAMPLE="${SAMPLE:-GE11174}"
OUTDIR="${OUTDIR:-resistome_virulence_${SAMPLE}}"
THREADS="${THREADS:-16}"
# ABRicate thresholds
# If you want your earlier "35 genes" behavior, use MINID=70 MINCOV=50.
# If you want stricter: e.g. MINID=80 MINCOV=70.
MINID="${MINID:-70}"
MINCOV="${MINCOV:-50}"
# -----------------------------------------------------
ts(){ date +"%F %T"; }
log(){ echo "[$(ts)] $*" >&2; }
on_err(){
local ec=$?
log "ERROR: failed (exit=${ec}) at line ${BASH_LINENO[0]}: ${BASH_COMMAND}"
exit $ec
}
trap on_err ERR
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
activate_env() {
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
conda activate "${ENV_NAME}"
}
main(){
activate_env
log "Env: ${ENV_NAME}"
log "ASM: ${ASM}"
log "Sample: ${SAMPLE}"
log "Outdir: ${OUTDIR}"
log "Threads: ${THREADS}"
log "ABRicate thresholds: MINID=${MINID} MINCOV=${MINCOV}"
mkdir -p "${OUTDIR}"/{raw,logs}
# Save full log
LOGFILE="${OUTDIR}/logs/run_$(date +'%F_%H%M%S').log"
exec > >(tee -a "${LOGFILE}") 2>&1
log "Tool versions:"
abricate --version || true
abricate-get_db --help | head -n 5 || true
log "ABRicate DB list (selected):"
abricate --list | egrep -i "vfdb|resfinder|megares|card" || true
log "Indexing ABRicate databases (safe to re-run)..."
abricate --setupdb
# ---------------- Run ABRicate ----------------
log "Running ABRicate: MEGARes"
abricate --db megares --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.megares.tab"
log "Running ABRicate: CARD"
abricate --db card --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.card.tab"
log "Running ABRicate: ResFinder"
abricate --db resfinder --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
log "Running ABRicate: VFDB"
abricate --db vfdb --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
# --------------- Build tables -----------------
export OUTDIR SAMPLE
export MEGARES_TAB="${OUTDIR}/raw/${SAMPLE}.megares.tab"
export CARD_TAB="${OUTDIR}/raw/${SAMPLE}.card.tab"
export RESFINDER_TAB="${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
export VFDB_TAB="${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
export AMR_OUT="${OUTDIR}/Table_AMR_genes_one_per_gene.tsv"
export VIR_OUT="${OUTDIR}/Table_Virulence_VFDB_dedup.tsv"
export STATUS_OUT="${OUTDIR}/Table_DB_hit_counts.tsv"
log "Generating deduplicated tables..."
python - <<'PY'
import os
import pandas as pd
from pathlib import Path
megares_tab = Path(os.environ["MEGARES_TAB"])
card_tab = Path(os.environ["CARD_TAB"])
resfinder_tab = Path(os.environ["RESFINDER_TAB"])
vfdb_tab = Path(os.environ["VFDB_TAB"])
amr_out = Path(os.environ["AMR_OUT"])
vir_out = Path(os.environ["VIR_OUT"])
status_out = Path(os.environ["STATUS_OUT"])
def read_abricate(path: Path) -> pd.DataFrame:
"""Parse ABRicate .tab where header line starts with '#FILE'."""
if (not path.exists()) or path.stat().st_size == 0:
return pd.DataFrame()
header = None
rows = []
with path.open("r", errors="replace") as f:
for line in f:
if not line.strip():
continue
if line.startswith("#FILE"):
header = line.lstrip("#").rstrip("\n").split("\t")
continue
if line.startswith("#"):
continue
rows.append(line.rstrip("\n").split("\t"))
if header is None:
return pd.DataFrame()
if not rows:
return pd.DataFrame(columns=header)
return pd.DataFrame(rows, columns=header)
def normalize(df: pd.DataFrame, dbname: str) -> pd.DataFrame:
cols_out = ["Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"]
if df is None or df.empty:
return pd.DataFrame(columns=cols_out)
out = pd.DataFrame({
"Database": dbname,
"Gene": df.get("GENE",""),
"Product": df.get("PRODUCT",""),
"Accession": df.get("ACCESSION",""),
"Contig": df.get("SEQUENCE",""),
"Start": df.get("START",""),
"End": df.get("END",""),
"Strand": df.get("STRAND",""),
"Pct_Identity": pd.to_numeric(df.get("%IDENTITY",""), errors="coerce"),
"Pct_Coverage": pd.to_numeric(df.get("%COVERAGE",""), errors="coerce"),
})
return out[cols_out]
def best_hit_dedup(df: pd.DataFrame, key_cols):
"""Keep best hit by highest identity, then coverage, then alignment length."""
if df.empty:
return df
d = df.copy()
d["Start_i"] = pd.to_numeric(d["Start"], errors="coerce").fillna(0).astype(int)
d["End_i"] = pd.to_numeric(d["End"], errors="coerce").fillna(0).astype(int)
d["Len"] = (d["End_i"] - d["Start_i"]).abs() + 1
d = d.sort_values(["Pct_Identity","Pct_Coverage","Len"], ascending=[False,False,False])
d = d.drop_duplicates(subset=key_cols, keep="first")
return d.drop(columns=["Start_i","End_i","Len"])
def count_hits(path: Path) -> int:
if not path.exists():
return 0
n = 0
with path.open() as f:
for line in f:
if line.startswith("#") or not line.strip():
continue
n += 1
return n
# -------- load + normalize --------
parts = []
for dbname, p in [("MEGARes", megares_tab), ("CARD", card_tab), ("ResFinder", resfinder_tab)]:
df = read_abricate(p)
parts.append(normalize(df, dbname))
amr_all = pd.concat([x for x in parts if not x.empty], ignore_index=True) if any(not x.empty for x in parts) else pd.DataFrame(
columns=["Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"]
)
# remove empty genes
amr_all = amr_all[amr_all["Gene"].astype(str).str.len() > 0].copy()
# best per (Database,Gene)
amr_db_gene = best_hit_dedup(amr_all, ["Database","Gene"]) if not amr_all.empty else amr_all
# one row per Gene overall, priority: CARD > ResFinder > MEGARes
priority = {"CARD": 0, "ResFinder": 1, "MEGARes": 2}
if not amr_db_gene.empty:
amr_db_gene["prio"] = amr_db_gene["Database"].map(priority).fillna(9).astype(int)
amr_one = amr_db_gene.sort_values(
["Gene","prio","Pct_Identity","Pct_Coverage"],
ascending=[True, True, False, False]
)
amr_one = amr_one.drop_duplicates(["Gene"], keep="first").drop(columns=["prio"])
amr_one = amr_one.sort_values(["Gene"]).reset_index(drop=True)
else:
amr_one = amr_db_gene
amr_out.parent.mkdir(parents=True, exist_ok=True)
amr_one.to_csv(amr_out, sep="\t", index=False)
# -------- VFDB --------
vf = normalize(read_abricate(vfdb_tab), "VFDB")
vf = vf[vf["Gene"].astype(str).str.len() > 0].copy()
vf_one = best_hit_dedup(vf, ["Gene"]) if not vf.empty else vf
if not vf_one.empty:
vf_one = vf_one.sort_values(["Gene"]).reset_index(drop=True)
vir_out.parent.mkdir(parents=True, exist_ok=True)
vf_one.to_csv(vir_out, sep="\t", index=False)
# -------- status counts --------
status = pd.DataFrame([
{"Database":"MEGARes", "Hit_lines": count_hits(megares_tab), "File": str(megares_tab)},
{"Database":"CARD", "Hit_lines": count_hits(card_tab), "File": str(card_tab)},
{"Database":"ResFinder", "Hit_lines": count_hits(resfinder_tab), "File": str(resfinder_tab)},
{"Database":"VFDB", "Hit_lines": count_hits(vfdb_tab), "File": str(vfdb_tab)},
])
status_out.parent.mkdir(parents=True, exist_ok=True)
status.to_csv(status_out, sep="\t", index=False)
print("OK wrote:")
print(" ", amr_out, "rows=", len(amr_one))
print(" ", vir_out, "rows=", len(vf_one))
print(" ", status_out)
PY
log "Finished."
log "Main outputs:"
log " ${AMR_OUT}"
log " ${VIR_OUT}"
log " ${STATUS_OUT}"
log "Raw ABRicate outputs:"
log " ${OUTDIR}/raw/${SAMPLE}.megares.tab"
log " ${OUTDIR}/raw/${SAMPLE}.card.tab"
log " ${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
log " ${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
log "Log:"
log " ${LOGFILE}"
}
mainresolve_best_assemblies_entrez.py#!/usr/bin/env python3
import csv
import os
import re
import sys
import time
from dataclasses import dataclass
from typing import List, Optional, Tuple
from Bio import Entrez
# REQUIRED by NCBI policy
Entrez.email = os.environ.get("NCBI_EMAIL", "your.email@example.com")
# Be nice to NCBI
ENTREZ_DELAY_SEC = float(os.environ.get("ENTREZ_DELAY_SEC", "0.34"))
LEVEL_RANK = {
"Complete Genome": 0,
"Chromosome": 1,
"Scaffold": 2,
"Contig": 3,
# sometimes NCBI uses slightly different strings:
"complete genome": 0,
"chromosome": 1,
"scaffold": 2,
"contig": 3,
}
def level_rank(level: str) -> int:
return LEVEL_RANK.get(level.strip(), 99)
def is_refseq(accession: str) -> bool:
return accession.startswith("GCF_")
@dataclass
class AssemblyHit:
assembly_uid: str
assembly_accession: str # GCF_... or GCA_...
organism: str
strain: str
assembly_level: str
refseq_category: str
submitter: str
ftp_path: str
def entrez_search_assembly(term: str, retmax: int = 50) -> List[str]:
"""Return Assembly UIDs matching term."""
h = Entrez.esearch(db="assembly", term=term, retmax=str(retmax))
rec = Entrez.read(h)
h.close()
time.sleep(ENTREZ_DELAY_SEC)
return rec.get("IdList", [])
def entrez_esummary_assembly(uids: List[str]) -> List[AssemblyHit]:
"""Fetch assembly summary records for given UIDs."""
if not uids:
return []
h = Entrez.esummary(db="assembly", id=",".join(uids), report="full")
rec = Entrez.read(h)
h.close()
time.sleep(ENTREZ_DELAY_SEC)
hits: List[AssemblyHit] = []
docs = rec.get("DocumentSummarySet", {}).get("DocumentSummary", [])
for d in docs:
# Some fields can be missing
acc = str(d.get("AssemblyAccession", "")).strip()
org = str(d.get("Organism", "")).strip()
level = str(d.get("AssemblyStatus", "")).strip() or str(d.get("AssemblyLevel", "")).strip()
# NCBI uses "AssemblyStatus" sometimes, "AssemblyLevel" other times;
# in practice AssemblyStatus often equals "Complete Genome"/"Chromosome"/...
if not level:
level = str(d.get("AssemblyLevel", "")).strip()
strain = str(d.get("Biosample", "")).strip()
# Strain is not always in a clean field. Try "Sub_value" in Meta, or parse Submitter/Title.
# We'll try a few common places:
title = str(d.get("AssemblyName", "")).strip()
submitter = str(d.get("SubmitterOrganization", "")).strip()
refcat = str(d.get("RefSeq_category", "")).strip()
ftp = str(d.get("FtpPath_RefSeq", "")).strip() or str(d.get("FtpPath_GenBank", "")).strip()
hits.append(
AssemblyHit(
assembly_uid=str(d.get("Uid", "")),
assembly_accession=acc,
organism=org,
strain=strain,
assembly_level=level,
refseq_category=refcat,
submitter=submitter,
ftp_path=ftp,
)
)
return hits
def best_hit(hits: List[AssemblyHit]) -> Optional[AssemblyHit]:
"""Pick best hit by level (Complete>Chromosome>...), prefer RefSeq, then prefer representative/reference."""
if not hits:
return None
def key(h: AssemblyHit) -> Tuple[int, int, int, str]:
# lower is better
lvl = level_rank(h.assembly_level)
ref = 0 if is_refseq(h.assembly_accession) else 1
# prefer reference/representative if present
cat = (h.refseq_category or "").lower()
rep = 0
if "reference" in cat:
rep = 0
elif "representative" in cat:
rep = 1
else:
rep = 2
# tie-breaker: accession string (stable)
return (lvl, ref, rep, h.assembly_accession)
return sorted(hits, key=key)[0]
def relaxed_fallback_terms(organism: str, strain_tokens: List[str]) -> List[str]:
"""
Build fallback search terms:
1) organism + strain tokens
2) organism only (species-only)
3) genus-only (if species fails)
"""
terms = []
# 1) Full term: organism + strain tokens
if strain_tokens:
t = f'"{organism}"[Organism] AND (' + " OR ".join(f'"{s}"[All Fields]' for s in strain_tokens) + ")"
terms.append(t)
# 2) Species only
terms.append(f'"{organism}"[Organism]')
# 3) Genus only
genus = organism.split()[0]
terms.append(f'"{genus}"[Organism]')
return terms
def resolve_one(label: str, organism: str, strain_tokens: List[str], retmax: int = 80) -> Tuple[str, Optional[AssemblyHit], str]:
"""
Returns:
- selected accession or "NA"
- selected hit (optional)
- which query term matched
"""
for term in relaxed_fallback_terms(organism, strain_tokens):
uids = entrez_search_assembly(term, retmax=retmax)
hits = entrez_esummary_assembly(uids)
chosen = best_hit(hits)
if chosen and chosen.assembly_accession:
return chosen.assembly_accession, chosen, term
return "NA", None, ""
def parse_targets_tsv(path: str) -> List[Tuple[str, str, List[str]]]:
"""
Input TSV format:
label organism strain_tokens
where strain_tokens is a semicolon-separated list, e.g. "FRB97;FRB 97"
"""
rows = []
with open(path, newline="") as f:
r = csv.DictReader(f, delimiter="\t")
for row in r:
label = row["label"].strip()
org = row["organism"].strip()
tokens = [x.strip() for x in row.get("strain_tokens", "").split(";") if x.strip()]
rows.append((label, org, tokens))
return rows
def main():
if len(sys.argv) < 3:
print("Usage: resolve_best_assemblies_entrez.py targets.tsv out.tsv", file=sys.stderr)
sys.exit(2)
targets_tsv = sys.argv[1]
out_tsv = sys.argv[2]
targets = parse_targets_tsv(targets_tsv)
with open(out_tsv, "w", newline="") as f:
w = csv.writer(f, delimiter="\t")
w.writerow(["label", "best_accession", "assembly_level", "refseq_category", "organism", "query_used"])
for label, org, tokens in targets:
acc, hit, term = resolve_one(label, org, tokens)
if hit:
w.writerow([label, acc, hit.assembly_level, hit.refseq_category, hit.organism, term])
print(f"[OK] {label} -> {acc} ({hit.assembly_level})")
else:
w.writerow([label, "NA", "", "", org, ""])
print(f"[WARN] {label} -> NA (no assemblies found)")
if __name__ == "__main__":
main()build_wgs_tree_fig3B.sh#!/usr/bin/env bash
set -euo pipefail
###############################################################################
# Core-genome phylogeny pipeline (genome-wide; no 16S/MLST):
#
# Uses existing conda env prefix:
# ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3
#
# Inputs:
# - resolved_accessions.tsv
# - REF.fasta
#
# Also consider these 4 accessions (duplicates removed):
# GCF_002291425.1, GCF_047901425.1, GCF_004342245.1, GCA_032062225.1
#
# Robustness:
# - Conda activation hook may reference JAVA_HOME under set -u (handled)
# - GFF validation ignores the ##FASTA FASTA block (valid GFF3)
# - FIXED: No more double Roary directories (script no longer pre-creates -f dir)
# Logs go to WORKDIR/logs and are also copied into the final Roary dir.
#
# Outputs:
# ${WORKDIR}/plot/core_tree.pdf
# ${WORKDIR}/plot/core_tree.png
###############################################################################
THREADS="${THREADS:-8}"
WORKDIR="${WORKDIR:-work_wgs_tree}"
RESOLVED_TSV="${RESOLVED_TSV:-resolved_accessions.tsv}"
REF_FASTA="${REF_FASTA:-AN6.fasta}"
ENV_NAME="${ENV_NAME:-/home/jhuang/miniconda3/envs/bengal3_ac3}"
EXTRA_ASSEMBLIES=(
#"GCF_002291425.1"
#"GCF_047901425.1"
#"GCF_004342245.1"
#"GCA_032062225.1"
)
CLUSTERS_K="${CLUSTERS_K:-6}"
MODE="${1:-all}"
log(){ echo "[$(date +'%F %T')] $*" >&2; }
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
activate_existing_env(){
if [[ ! -d "${ENV_NAME}" ]]; then
log "ERROR: ENV_NAME path does not exist: ${ENV_NAME}"
exit 1
fi
conda_base="$(dirname "$(dirname "${ENV_NAME}")")"
if [[ -f "${conda_base}/etc/profile.d/conda.sh" ]]; then
# shellcheck disable=SC1091
source "${conda_base}/etc/profile.d/conda.sh"
else
if need_cmd conda; then
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
else
log "ERROR: cannot find conda.sh and conda is not on PATH."
exit 1
fi
fi
# Avoid "unbound variable" in activation hooks under set -u
export JAVA_HOME="${JAVA_HOME:-}"
log "Activating env: ${ENV_NAME}"
set +u
conda activate "${ENV_NAME}"
set -u
}
check_dependencies() {
# ---- plot-only mode: only need R (and optionally python) ----
if [[ "${MODE}" == "plot-only" ]]; then
local missing=()
command -v Rscript >/dev/null 2>&1 || missing+=("Rscript")
command -v python >/dev/null 2>&1 || missing+=("python")
if (( ${#missing[@]} )); then
log "ERROR: Missing required tools for plot-only in env: ${ENV_NAME}"
printf ' - %s\n' "${missing[@]}" >&2
exit 1
fi
# Check required R packages (fail early with clear message)
Rscript -e 'pkgs <- c("ggtree","ggplot2","aplot");
miss <- pkgs[!sapply(pkgs, requireNamespace, quietly=TRUE)];
if(length(miss)) stop("Missing R packages: ", paste(miss, collapse=", "))'
return 0
fi
# ------------------------------------------------------------
# existing full-pipeline checks continue below...
# (your current prokka/roary/raxml-ng checks stay as-is)
#...
}
prepare_accessions(){
[[ -s "${RESOLVED_TSV}" ]] || { log "ERROR: missing ${RESOLVED_TSV}"; exit 1; }
mkdir -p "${WORKDIR}/meta"
printf "%s\n" "${EXTRA_ASSEMBLIES[@]}" > "${WORKDIR}/meta/extras.txt"
WORKDIR="${WORKDIR}" RESOLVED_TSV="${RESOLVED_TSV}" python - << 'PY'
import os
import pandas as pd
import pathlib
workdir = pathlib.Path(os.environ.get("WORKDIR", "work_wgs_tree"))
resolved_tsv = os.environ.get("RESOLVED_TSV", "resolved_accessions.tsv")
df = pd.read_csv(resolved_tsv, sep="\t")
# Expect columns like: label, best_accession (but be tolerant)
if "best_accession" not in df.columns:
df = df.rename(columns={df.columns[1]:"best_accession"})
if "label" not in df.columns:
df = df.rename(columns={df.columns[0]:"label"})
df = df[["label","best_accession"]].dropna()
df = df[df["best_accession"]!="NA"].copy()
extras_path = workdir/"meta/extras.txt"
extras = [x.strip() for x in extras_path.read_text().splitlines() if x.strip()]
extra_df = pd.DataFrame({"label":[f"EXTRA_{a}" for a in extras], "best_accession": extras})
all_df = pd.concat([df, extra_df], ignore_index=True)
all_df = all_df.drop_duplicates(subset=["best_accession"], keep="first").reset_index(drop=True)
out = workdir/"meta/accessions.tsv"
out.parent.mkdir(parents=True, exist_ok=True)
all_df.to_csv(out, sep="\t", index=False)
print("Final unique genomes:", len(all_df))
print(all_df)
print("Wrote:", out)
PY
}
download_genomes(){
mkdir -p "${WORKDIR}/genomes_ncbi"
while IFS=$'\t' read -r label acc; do
[[ "$label" == "label" ]] && continue
[[ -z "${acc}" ]] && continue
outdir="${WORKDIR}/genomes_ncbi/${acc}"
if [[ -d "${outdir}" ]]; then
log "Found ${acc}, skipping download"
continue
fi
log "Downloading ${acc}..."
datasets download genome accession "${acc}" --include genome --filename "${WORKDIR}/genomes_ncbi/${acc}.zip"
unzip -q "${WORKDIR}/genomes_ncbi/${acc}.zip" -d "${outdir}"
rm -f "${WORKDIR}/genomes_ncbi/${acc}.zip"
done < "${WORKDIR}/meta/accessions.tsv"
}
collect_fastas(){
mkdir -p "${WORKDIR}/fastas"
while IFS=$'\t' read -r label acc; do
[[ "$label" == "label" ]] && continue
[[ -z "${acc}" ]] && continue
fna="$(find "${WORKDIR}/genomes_ncbi/${acc}" -type f -name "*.fna" | head -n 1 || true)"
[[ -n "${fna}" ]] || { log "ERROR: .fna not found for ${acc}"; exit 1; }
cp -f "${fna}" "${WORKDIR}/fastas/${acc}.fna"
done < "${WORKDIR}/meta/accessions.tsv"
[[ -s "${REF_FASTA}" ]] || { log "ERROR: missing ${REF_FASTA}"; exit 1; }
cp -f "${REF_FASTA}" "${WORKDIR}/fastas/REF.fna"
}
run_prokka(){
mkdir -p "${WORKDIR}/prokka" "${WORKDIR}/gffs"
for fna in "${WORKDIR}/fastas/"*.fna; do
base="$(basename "${fna}" .fna)"
outdir="${WORKDIR}/prokka/${base}"
gffout="${WORKDIR}/gffs/${base}.gff"
if [[ -s "${gffout}" ]]; then
log "GFF exists for ${base}, skipping Prokka"
continue
fi
log "Prokka annotating ${base}..."
prokka --outdir "${outdir}" --prefix "${base}" --cpus "${THREADS}" --force "${fna}"
cp -f "${outdir}/${base}.gff" "${gffout}"
done
}
sanitize_and_check_gffs(){
log "Sanity checking GFFs (ignoring ##FASTA section)..."
for gff in "${WORKDIR}/gffs/"*.gff; do
if file "$gff" | grep -qi "CRLF"; then
log "Fixing CRLF -> LF in $(basename "$gff")"
sed -i 's/\r$//' "$gff"
fi
bad=$(awk '
BEGIN{bad=0; in_fasta=0}
/^##FASTA/{in_fasta=1; next}
in_fasta==1{next}
/^#/{next}
NF==0{next}
{
if (split($0,a,"\t")!=9) {bad=1}
}
END{print bad}
' "$gff")
if [[ "$bad" == "1" ]]; then
log "ERROR: GFF feature section not 9-column tab-delimited: $gff"
log "First 5 problematic feature lines (before ##FASTA):"
awk '
BEGIN{in_fasta=0; c=0}
/^##FASTA/{in_fasta=1; next}
in_fasta==1{next}
/^#/{next}
NF==0{next}
{
if (split($0,a,"\t")!=9) {
print
c++
if (c==5) exit
}
}
' "$gff" || true
exit 1
fi
done
}
run_roary(){
mkdir -p "${WORKDIR}/meta" "${WORKDIR}/logs"
ts="$(date +%s)"
run_id="${ts}_$$"
ROARY_OUT="${WORKDIR}/roary_${run_id}"
ROARY_STDOUT="${WORKDIR}/logs/roary_${run_id}.stdout.txt"
ROARY_STDERR="${WORKDIR}/logs/roary_${run_id}.stderr.txt"
MARKER="${WORKDIR}/meta/roary_${run_id}.start"
: > "${MARKER}"
log "Running Roary (outdir: ${ROARY_OUT})"
log "Roary logs:"
log " STDOUT: ${ROARY_STDOUT}"
log " STDERR: ${ROARY_STDERR}"
set +e
roary -e --mafft -p "${THREADS}" -cd 95 -i 95 \
-f "${ROARY_OUT}" "${WORKDIR}/gffs/"*.gff \
> "${ROARY_STDOUT}" 2> "${ROARY_STDERR}"
rc=$?
set -e
if [[ "${rc}" -ne 0 ]]; then
log "WARNING: Roary exited non-zero (rc=${rc}). Will check if core alignment was produced anyway."
fi
CORE_ALN="$(find "${WORKDIR}" -maxdepth 2 -type f -name "core_gene_alignment.aln" -newer "${MARKER}" -printf '%T@ %p\n' 2>/dev/null \
| sort -nr | head -n 1 | cut -d' ' -f2- || true)"
if [[ -z "${CORE_ALN}" || ! -s "${CORE_ALN}" ]]; then
log "ERROR: Could not find core_gene_alignment.aln produced by this Roary run under ${WORKDIR}"
log "---- STDERR (head) ----"
head -n 120 "${ROARY_STDERR}" 2>/dev/null || true
log "---- STDERR (tail) ----"
tail -n 120 "${ROARY_STDERR}" 2>/dev/null || true
exit 1
fi
CORE_DIR="$(dirname "${CORE_ALN}")"
cp -f "${ROARY_STDOUT}" "${CORE_DIR}/roary.stdout.txt" || true
cp -f "${ROARY_STDERR}" "${CORE_DIR}/roary.stderr.txt" || true
# >>> IMPORTANT FIX: store ABSOLUTE path <<<
CORE_ALN_ABS="$(readlink -f "${CORE_ALN}")"
log "Using core alignment: ${CORE_ALN_ABS}"
echo "${CORE_ALN_ABS}" > "${WORKDIR}/meta/core_alignment_path.txt"
echo "$(readlink -f "${CORE_DIR}")" > "${WORKDIR}/meta/roary_output_dir.txt"
}
run_raxmlng(){
mkdir -p "${WORKDIR}/raxmlng"
CORE_ALN="$(cat "${WORKDIR}/meta/core_alignment_path.txt")"
[[ -s "${CORE_ALN}" ]] || { log "ERROR: core alignment not found or empty: ${CORE_ALN}"; exit 1; }
log "Running RAxML-NG..."
raxml-ng --all \
--msa "${CORE_ALN}" \
--model GTR+G \
--bs-trees 1000 \
--threads "${THREADS}" \
--prefix "${WORKDIR}/raxmlng/core"
}
ensure_r_pkgs(){
Rscript - <<'RS'
need <- c("ape","ggplot2","dplyr","readr","aplot","ggtree")
missing <- need[!vapply(need, requireNamespace, logical(1), quietly=TRUE)]
if (length(missing)) {
message("Missing R packages: ", paste(missing, collapse=", "))
message("Try:")
message(" conda install -c conda-forge -c bioconda r-aplot bioconductor-ggtree r-ape r-ggplot2 r-dplyr r-readr")
quit(status=1)
}
RS
}
plot_tree(){
mkdir -p "${WORKDIR}/plot"
WORKDIR="${WORKDIR}" python - << 'PY'
import os
import pandas as pd
import pathlib
workdir = pathlib.Path(os.environ.get("WORKDIR", "work_wgs_tree"))
acc = pd.read_csv(workdir/"meta/accessions.tsv", sep="\t")
g = (acc.groupby("best_accession")["label"]
.apply(lambda x: "; ".join(sorted(set(map(str, x)))))
.reset_index())
g["display"] = g.apply(lambda r: f'{r["label"]} ({r["best_accession"]})', axis=1)
labels = g.rename(columns={"best_accession":"sample"})[["sample","display"]]
# Add REF
labels = pd.concat([labels, pd.DataFrame([{"sample":"REF","display":"REF"}])], ignore_index=True)
out = workdir/"plot/labels.tsv"
out.parent.mkdir(parents=True, exist_ok=True)
labels.to_csv(out, sep="\t", index=False)
print("Wrote:", out)
PY
cat > "${WORKDIR}/plot/plot_tree.R" << 'RS'
suppressPackageStartupMessages({
library(ape); library(ggplot2); library(ggtree); library(dplyr); library(readr)
})
args <- commandArgs(trailingOnly=TRUE)
tree_in <- args[1]; labels_tsv <- args[2]; k <- as.integer(args[3])
out_pdf <- args[4]; out_png <- args[5]
tr <- read.tree(tree_in)
lab <- read_tsv(labels_tsv, show_col_types=FALSE)
tipmap <- setNames(lab$display, lab$sample)
tr$tip.label <- ifelse(tr$tip.label %in% names(tipmap), tipmap[tr$tip.label], tr$tip.label)
hc <- as.hclust.phylo(tr)
grp <- cutree(hc, k=k)
grp_df <- tibble(tip=names(grp), clade=paste0("Clade_", grp))
p <- ggtree(tr, layout="rectangular") %<+% grp_df +
aes(color=clade) +
geom_tree(linewidth=0.9) +
geom_tippoint(aes(color=clade), size=2.3) +
geom_tiplab(aes(color=clade), size=3.1, align=TRUE,
linetype="dotted", linesize=0.35, offset=0.02) +
theme_tree2() +
theme(legend.position="right", legend.title=element_blank(),
plot.margin=margin(8,18,8,8))
# + geom_treescale(x=0, y=0, width=0.01, fontsize=3)
# ---- Manual scale bar (fixed label "0.01") ----
scale_x <- 0
scale_y <- 0
scale_w <- 0.01
p <- p +
annotate("segment",
x = scale_x, xend = scale_x + scale_w,
y = scale_y, yend = scale_y,
linewidth = 0.6) +
annotate("text",
x = scale_x + scale_w/2,
y = scale_y - 0.6,
label = "0.01",
size = 3)
# ----------------------------------------------
ggsave(out_pdf, p, width=9, height=6.5, device="pdf")
ggsave(out_png, p, width=9, height=6.5, dpi=300)
RS
Rscript "${WORKDIR}/plot/plot_tree.R" \
"${WORKDIR}/raxmlng/core.raxml.support" \
"${WORKDIR}/plot/labels.tsv" \
"${CLUSTERS_K}" \
"${WORKDIR}/plot/core_tree.pdf" \
"${WORKDIR}/plot/core_tree.png"
log "Plot written:"
log " ${WORKDIR}/plot/core_tree.pdf"
log " ${WORKDIR}/plot/core_tree.png"
}
main(){
mkdir -p "${WORKDIR}"
activate_existing_env
check_dependencies
if [[ "${MODE}" == "plot-only" ]]; then
log "Running plot-only mode"
plot_tree
log "DONE."
exit 0
fi
log "1) Prepare unique accessions"
prepare_accessions
log "2) Download genomes"
download_genomes
log "3) Collect FASTAs (+ REF)"
collect_fastas
log "4) Prokka"
run_prokka
log "4b) GFF sanity check"
sanitize_and_check_gffs
log "5) Roary"
run_roary
log "6) RAxML-NG"
run_raxmlng
#log "6b) Check R packages"
#ensure_r_pkgs
log "7) Plot"
plot_tree
log "DONE."
}
main "$@"regenerate_labels.shpython - <<'PY'
import json, re
from pathlib import Path
import pandas as pd
WORKDIR = Path("work_wgs_tree")
ACC_TSV = WORKDIR / "meta/accessions.tsv"
GENOMES_DIR = WORKDIR / "genomes_ncbi"
OUT = WORKDIR / "plot/labels.tsv"
def first_existing(paths):
for p in paths:
if p and Path(p).exists():
return Path(p)
return None
def find_metadata_files(acc_dir: Path):
# NCBI Datasets layouts vary by version; search broadly
candidates = []
for pat in [
"**/assembly_data_report.jsonl",
"**/data_report.jsonl",
"**/dataset_catalog.json",
"**/*assembly_report*.txt",
"**/*assembly_report*.tsv",
]:
candidates += list(acc_dir.glob(pat))
# de-dup, keep stable order
seen = set()
uniq = []
for p in candidates:
if p.as_posix() not in seen:
uniq.append(p)
seen.add(p.as_posix())
return uniq
def parse_jsonl_for_name_and_strain(p: Path):
# assembly_data_report.jsonl / data_report.jsonl: first JSON object usually has organism info
try:
with p.open() as f:
for line in f:
line = line.strip()
if not line:
continue
obj = json.loads(line)
# Try common fields
# organismName may appear as:
# obj["organism"]["organismName"] or obj["organismName"]
org = None
strain = None
if isinstance(obj, dict):
if "organism" in obj and isinstance(obj["organism"], dict):
org = obj["organism"].get("organismName") or obj["organism"].get("taxName")
# isolate/strain can hide in infraspecificNames or isolate/strain keys
infra = obj["organism"].get("infraspecificNames") or {}
if isinstance(infra, dict):
strain = infra.get("strain") or infra.get("isolate")
strain = strain or obj["organism"].get("strain") or obj["organism"].get("isolate")
org = org or obj.get("organismName") or obj.get("taxName")
# Sometimes isolate/strain is nested elsewhere
if not strain:
# assemblyInfo / assembly / sampleInfo patterns
for key in ["assemblyInfo", "assembly", "sampleInfo", "biosample"]:
if key in obj and isinstance(obj[key], dict):
d = obj[key]
strain = strain or d.get("strain") or d.get("isolate")
infra = d.get("infraspecificNames")
if isinstance(infra, dict):
strain = strain or infra.get("strain") or infra.get("isolate")
if org:
return org, strain
except Exception:
pass
return None, None
def parse_dataset_catalog(p: Path):
# dataset_catalog.json can include assembly/organism info, but structure varies.
try:
obj = json.loads(p.read_text())
except Exception:
return None, None
org = None
strain = None
# walk dict recursively looking for likely keys
def walk(x):
nonlocal org, strain
if isinstance(x, dict):
# organism keys
if not org:
if "organismName" in x and isinstance(x["organismName"], str):
org = x["organismName"]
elif "taxName" in x and isinstance(x["taxName"], str):
org = x["taxName"]
# strain/isolate keys
if not strain:
for k in ["strain", "isolate"]:
if k in x and isinstance(x[k], str) and x[k].strip():
strain = x[k].strip()
break
for v in x.values():
walk(v)
elif isinstance(x, list):
for v in x:
walk(v)
walk(obj)
return org, strain
def parse_assembly_report_txt(p: Path):
# NCBI assembly_report.txt often has lines like: "# Organism name:" and "# Infraspecific name:"
org = None
strain = None
try:
for line in p.read_text(errors="ignore").splitlines():
if line.startswith("# Organism name:"):
org = line.split(":", 1)[1].strip()
elif line.startswith("# Infraspecific name:"):
val = line.split(":", 1)[1].strip()
# e.g. "strain=XXXX" or "isolate=YYYY"
m = re.search(r"(strain|isolate)\s*=\s*(.+)", val)
if m:
strain = m.group(2).strip()
if org and strain:
break
except Exception:
pass
return org, strain
def best_name_for_accession(acc: str):
acc_dir = GENOMES_DIR / acc
if not acc_dir.exists():
return None
files = find_metadata_files(acc_dir)
org = None
strain = None
# Prefer JSONL reports first
for p in files:
if p.name.endswith(".jsonl"):
org, strain = parse_jsonl_for_name_and_strain(p)
if org:
break
# Next try dataset_catalog.json
if not org:
for p in files:
if p.name == "dataset_catalog.json":
org, strain = parse_dataset_catalog(p)
if org:
break
# Finally try assembly report text
if not org:
for p in files:
if "assembly_report" in p.name and p.suffix in [".txt", ".tsv"]:
org, strain = parse_assembly_report_txt(p)
if org:
break
if not org:
return None
# normalize whitespace
org = re.sub(r"\s+", " ", org).strip()
if strain:
strain = re.sub(r"\s+", " ", str(strain)).strip()
# avoid duplicating if strain already in organism string
if strain and strain.lower() not in org.lower():
return f"{org} {strain}"
return org
# --- build labels ---
acc = pd.read_csv(ACC_TSV, sep="\t")
if "label" not in acc.columns or "best_accession" not in acc.columns:
raise SystemExit("accessions.tsv must have columns: label, best_accession")
rows = []
for _, r in acc.iterrows():
label = str(r["label"])
accn = str(r["best_accession"])
if label.startswith("EXTRA_"):
nm = best_name_for_accession(accn)
if nm:
label = nm
else:
# fallback: keep previous behavior if metadata not found
label = label.replace("EXTRA_", "EXTRA ")
display = f"{label} ({accn})"
rows.append({"sample": accn, "display": display})
# Add GE11174 exactly as-is
rows.append({"sample": "GE11174", "display": "GE11174"})
out_df = pd.DataFrame(rows).drop_duplicates(subset=["sample"], keep="first")
OUT.parent.mkdir(parents=True, exist_ok=True)
out_df.to_csv(OUT, sep="\t", index=False)
print("Wrote:", OUT)
print(out_df)
PYplot_tree_v4.RsuppressPackageStartupMessages({
library(ape)
library(readr)
})
args <- commandArgs(trailingOnly = TRUE)
tree_in <- args[1]
labels_tsv <- args[2]
# args[3] is k (ignored here since all-black)
out_pdf <- args[4]
out_png <- args[5]
# --- Load tree ---
tr <- read.tree(tree_in)
# --- Root on outgroup (Brenneria nigrifluens) by accession ---
outgroup_id <- "GCF_005484965.1"
if (outgroup_id %in% tr$tip.label) {
tr <- root(tr, outgroup = outgroup_id, resolve.root = TRUE)
} else {
warning("Outgroup tip not found in tree: ", outgroup_id, " (tree will remain unrooted)")
}
# Make plotting order nicer
tr <- ladderize(tr, right = FALSE)
# --- Load labels (columns: sample, display) ---
lab <- read_tsv(labels_tsv, show_col_types = FALSE)
if (!all(c("sample","display") %in% colnames(lab))) {
stop("labels.tsv must contain columns: sample, display")
}
# Map tip labels AFTER rooting (rooting uses accession IDs)
tipmap <- setNames(lab$display, lab$sample)
tr$tip.label <- ifelse(tr$tip.label %in% names(tipmap),
unname(tipmap[tr$tip.label]),
tr$tip.label)
# --- Plot helper ---
plot_one <- function(device_fun) {
device_fun()
op <- par(no.readonly = TRUE)
on.exit(par(op), add = TRUE)
# Bigger right margin for long labels; tighter overall
par(mar = c(4, 2, 2, 18), xpd = NA)
# Compute xlim with padding so labels fit but whitespace is limited
xx <- node.depth.edgelength(tr)
xmax <- max(xx)
xpad <- 0.10 * xmax
plot(tr,
type = "phylogram",
use.edge.length = TRUE,
show.tip.label = TRUE,
edge.color = "black",
tip.color = "black",
cex = 0.9, # smaller text -> less overlap
label.offset = 0.003, # small gap after tip
no.margin = FALSE,
x.lim = c(0, xmax + xpad))
# Add a clear scale bar near bottom-left
# Use a fixed fraction of tree length for bar length
bar_len <- 0.05 * xmax
add.scale.bar(x = 0, y = 0, length = 0.01, lwd = 2, cex = 0.9)
}
# --- Write outputs (shorter height -> less vertical whitespace) ---
plot_one(function() pdf(out_pdf, width = 11, height = 6, useDingbats = FALSE))
dev.off()
plot_one(function() png(out_png, width = 3000, height = 1000, res = 300))
dev.off()
cat("Wrote:\n", out_pdf, "\n", out_png, "\n", sep = "")run_fastani_batch_verbose.sh#!/usr/bin/env bash
set -euo pipefail
# ============ CONFIG ============
QUERY="bacass_out/Prokka/An6/An6.fna" # 你的 query fasta
ACC_LIST="accessions.txt" # 每行一个 GCF/GCA
OUTDIR="fastani_batch"
THREADS=8
SUFFIX=".genomic.fna"
# =================================
ts() { date +"%F %T"; }
log() { echo "[$(ts)] $*"; }
die() { echo "[$(ts)] ERROR: $*" >&2; exit 1; }
# --- checks ---
log "Checking required commands..."
for cmd in fastANI awk sort unzip find grep wc head readlink; do
command -v "$cmd" >/dev/null 2>&1 || die "Missing command: $cmd"
done
command -v datasets >/dev/null 2>&1 || die "Missing NCBI datasets CLI. Install from NCBI Datasets."
[[ -f "$QUERY" ]] || die "QUERY not found: $QUERY"
[[ -f "$ACC_LIST" ]] || die "Accession list not found: $ACC_LIST"
log "QUERY: $QUERY"
log "ACC_LIST: $ACC_LIST"
log "OUTDIR: $OUTDIR"
log "THREADS: $THREADS"
mkdir -p "$OUTDIR/ref_fasta" "$OUTDIR/zips" "$OUTDIR/tmp" "$OUTDIR/logs"
REF_LIST="$OUTDIR/ref_list.txt"
QUERY_LIST="$OUTDIR/query_list.txt"
RAW_OUT="$OUTDIR/fastani_raw.tsv"
FINAL_OUT="$OUTDIR/fastani_results.tsv"
DL_LOG="$OUTDIR/logs/download.log"
ANI_LOG="$OUTDIR/logs/fastani.log"
: > "$REF_LIST"
: > "$DL_LOG"
: > "$ANI_LOG"
# --- build query list ---
q_abs="$(readlink -f "$QUERY")"
echo "$q_abs" > "$QUERY_LIST"
log "Wrote query list: $QUERY_LIST"
log " -> $q_abs"
# --- download refs ---
log "Downloading reference genomes via NCBI datasets..."
n_ok=0
n_skip=0
while read -r acc; do
[[ -z "$acc" ]] && continue
[[ "$acc" =~ ^# ]] && continue
log "Ref: $acc"
zip="$OUTDIR/zips/${acc}.zip"
unpack="$OUTDIR/tmp/$acc"
out_fna="$OUTDIR/ref_fasta/${acc}${SUFFIX}"
# download zip
log " - datasets download -> $zip"
if datasets download genome accession "$acc" --include genome --filename "$zip" >>"$DL_LOG" 2>&1; then
log " - download OK"
else
log " - download FAILED (see $DL_LOG), skipping $acc"
n_skip=$((n_skip+1))
continue
fi
# unzip
rm -rf "$unpack"
mkdir -p "$unpack"
log " - unzip -> $unpack"
if unzip -q "$zip" -d "$unpack" >>"$DL_LOG" 2>&1; then
log " - unzip OK"
else
log " - unzip FAILED (see $DL_LOG), skipping $acc"
n_skip=$((n_skip+1))
continue
fi
# find genomic.fna (兼容不同包结构:优先找 genomic.fna,其次找任何 .fna)
fna="$(find "$unpack" -type f \( -name "*genomic.fna" -o -name "*genomic.fna.gz" \) | head -n 1 || true)"
if [[ -z "${fna:-}" ]]; then
log " - genomic.fna not found, try any *.fna"
fna="$(find "$unpack" -type f -name "*.fna" | head -n 1 || true)"
fi
if [[ -z "${fna:-}" ]]; then
log " - FAILED to find any .fna in package (see $DL_LOG). skipping $acc"
n_skip=$((n_skip+1))
continue
fi
# handle gz if needed
if [[ "$fna" == *.gz ]]; then
log " - found gzipped fasta: $(basename "$fna"), gunzip -> $out_fna"
gunzip -c "$fna" > "$out_fna"
else
log " - found fasta: $(basename "$fna"), copy -> $out_fna"
cp -f "$fna" "$out_fna"
fi
# sanity check fasta looks non-empty
if [[ ! -s "$out_fna" ]]; then
log " - output fasta is empty, skipping $acc"
n_skip=$((n_skip+1))
continue
fi
echo "$(readlink -f "$out_fna")" >> "$REF_LIST"
n_ok=$((n_ok+1))
log " - saved ref fasta OK"
done < "$ACC_LIST"
log "Download summary: OK=$n_ok, skipped=$n_skip"
log "Ref list written: $REF_LIST ($(wc -l < "$REF_LIST") refs)"
if [[ "$(wc -l < "$REF_LIST")" -eq 0 ]]; then
die "No references available. Check $DL_LOG"
fi
# --- run fastANI ---
log "Running fastANI..."
log "Command:"
log " fastANI -ql $QUERY_LIST -rl $REF_LIST -t $THREADS -o $RAW_OUT"
# 重要:不要吞掉错误信息,把 stdout/stderr 进日志
if fastANI -ql "$QUERY_LIST" -rl "$REF_LIST" -t "$THREADS" -o "$RAW_OUT" >>"$ANI_LOG" 2>&1; then
log "fastANI finished (see $ANI_LOG)"
else
log "fastANI FAILED (see $ANI_LOG)"
die "fastANI failed. Inspect $ANI_LOG"
fi
# --- verify raw output ---
if [[ ! -f "$RAW_OUT" ]]; then
die "fastANI did not create $RAW_OUT. Check $ANI_LOG"
fi
if [[ ! -s "$RAW_OUT" ]]; then
die "fastANI output is empty ($RAW_OUT). Check $ANI_LOG; also verify fasta validity."
fi
log "fastANI raw output: $RAW_OUT ($(wc -l < "$RAW_OUT") lines)"
log "Sample lines:"
head -n 5 "$RAW_OUT" || true
# --- create final table ---
log "Creating final TSV with header..."
echo -e "Query\tReference\tANI\tMatchedFrag\tTotalFrag" > "$FINAL_OUT"
awk 'BEGIN{OFS="\t"} {print $1,$2,$3,$4,$5}' "$RAW_OUT" >> "$FINAL_OUT"
log "Final results: $FINAL_OUT ($(wc -l < "$FINAL_OUT") lines incl. header)"
log "Top hits (ANI desc):"
tail -n +2 "$FINAL_OUT" | sort -k3,3nr | head -n 10 || true
log "DONE."
log "Logs:"
log " download log: $DL_LOG"
log " fastANI log: $ANI_LOG"| Metrics* | Wildtype | ΔadeAB | ΔadeIJ | ΔcraA |
|---|---|---|---|---|
| Genome size (bp) | ||||
| Contig count | ||||
| Total number of reads sequenced | ||||
| Coverage depth (sequencing depth) | ||||
| Coarse consistency (%) | ||||
| Fine consistency (%) | ||||
| Completeness (%) | ||||
| Contamination (%) | ||||
| Contigs N50 (bp) | ||||
| Contigs L50 | ||||
| Guanine-cytosine content (%) | ||||
| Number of genes | ||||
| Number of coding sequences (CDSs) | ||||
| Number of tRNAs | ||||
| Number of rRNAs |
Legend / Footnote:
* Genome completeness represents the fraction of universal single-copy functional roles expected for a given taxonomic lineage that are detected in the genome; missing roles indicate incomplete genome assembly or annotation. Contamination is inferred from the detection of multiple copies of these roles, which are expected to be single-copy, suggesting potential contamination or strain heterogeneity. Coarse consistency measures whether the expected functional roles are present or absent as predicted, reflecting broad agreement in the genome annotation. Fine consistency measures whether the number of copies of each functional role matches what is expected based on the genome’s overall pattern, identifying small differences in gene counts. These metrics were calculated using subtools of the BV-BRC platform v3.51.7 (20): EvalG, which estimates genome completeness and contamination based on lineage-specific single-copy marker roles; and EvalCon, which assesses coarse and fine consistency using a machine-learning-derived catalog of approximately 1,300 functional roles with predictable relationships. Coverage depth (sequencing depth) refers to the average number of reads covering each base across the assembled genome, calculated as the total number of clean bases divided by the genome size.
💡 Note for your post: You can use this as the “Goal” section of your tutorial or reminder post, followed by the step-by-step command-line instructions (e.g., seqkit, checkm2, mosdepth, and Python parsing) that we developed to automatically fill in these exact blank fields.
The complete list of tools and services available on the platform (e.g., BV-BRC), organized into a clean, hierarchical, and highly readable format for easy reference:
💡 Tip for your workflow: If you need to extract the Coarse Consistency and Fine Consistency metrics for your manuscript table (as discussed earlier), you will primarily use the Genome Annotation or Comprehensive Genome Analysis (B) services under the Genomics category, as these trigger the EvalG and EvalCon subtools.
This is excellent news! Since you already have NCBI PGAP-annotated GenBank (.gb) files, we can completely skip Prokka. The GenBank COMMENT block contains the exact, high-quality annotation metrics (Genes, CDS, tRNA, rRNA, and Coverage) you need for your table.
Here is the updated, streamlined pipeline for Ubuntu. It uses your existing .fna files for assembly/QC metrics and the new .gb files for annotation metrics.
# Activate your conda environment (or install via mamba/conda if needed)
conda activate genome_stats
# Ensure you have seqkit and checkm2
mamba install -c bioconda seqkit checkm2 -yBased on your previous grep output and the new GenBank snippets, map your files. Navigate to your working directory:
cd ~/DATA/Data_Foong_DNAseq_2021_ATCC19606_Cm/bacass_out/checkm_input(Note: Ensure both the .fna and .gb files are in this directory, or adjust the paths in the Python script below).
Use seqkit on the .fna files to get the physical assembly statistics.
seqkit stats *.fna > assembly_stats.tsv
cat assembly_stats.tsvUse checkm2 on the .fna files (this remains the gold standard for these specific metrics).
# Run checkm2 prediction (adjust --threads based on your CPU cores)
checkm2 predict --input *.fna --output-directory checkm2_out --threads 8
# View the results
cat checkm2_out/quality_report.tsvThis updated Python script will:
seqkit and checkm2 outputs.COMMENT block from your .gb files to extract Coverage, Genes, CDS, tRNAs, and rRNAs.Reads = (Coverage × Genome Size) / (2 × Read_Length). (Assumes 150bp paired-end reads. Adjust READ_LENGTH in the script if your sequencing was 250bp).Save the following code as generate_table_from_gb.py and run it:
import os
import re
import pandas as pd
# 1. Map your specific filenames (adjust extensions if they differ in your folder)
files = {
"Wildtype": {
"fna": "A6WT_chr_plasmids.fna",
"gb": "A6WT_chr_plasmids.bgpipe.output_2799988.gb" # Adjust suffix if needed
},
"ΔadeAB": {
"fna": "adeAB_chr_plasmids.fna",
"gb": "adeAB_chr_plasmids.bgpipe.output_1954487.gb"
},
"ΔadeIJ": {
"fna": "adeIJ_chr_plasmids.fna",
"gb": "adeIJ_chr_plasmids.bgpipe.output_2028963.gb"
},
"ΔcraA": {
"fna": "A10CraA_chr_plasmids.fna",
"gb": "A10CraA_chr_plasmids.bgpipe.output_1118303.gb"
}
}
# Initialize table structure
metrics_list = [
"Genome size (bp)", "Contig count", "Total number of reads sequenced",
"Coverage depth (sequencing depth)", "Coarse consistency (%)", "Fine consistency (%)",
"Completeness (%)", "Contamination (%)", "Contigs N50 (bp)", "Contigs L50",
"Guanine-cytosine content (%)", "Number of genes", "Number of coding sequences (CDSs)",
"Number of tRNAs", "Number of rRNAs"
]
data = {metric: ["N/A"] * 4 for metric in metrics_list}
data["Metrics"] = metrics_list
def parse_gb_file(gb_path):
"""Extract annotation metrics directly from PGAP GenBank COMMENT block."""
if not os.path.exists(gb_path):
# Fallback: try to find any .gb file matching the base name
base = os.path.basename(gb_path).split('.bgpipe')[0]
matches = [f for f in os.listdir('.') if f.startswith(base) and f.endswith('.gb')]
gb_path = matches[0] if matches else gb_path
if not os.path.exists(gb_path):
return None
with open(gb_path, 'r', encoding='utf-8') as f:
content = f.read()
# Extract Coverage (e.g., "Genome Coverage :: 360x")
cov_match = re.search(r'Genome Coverage\s+::\s+([\d.]+)x', content)
coverage = cov_match.group(1) if cov_match else 'N/A'
# Extract Genes (total)
genes_match = re.search(r'Genes \(total\)\s+::\s+([\d,]+)', content)
genes = genes_match.group(1).replace(',', '') if genes_match else 'N/A'
# Extract CDSs (total)
cds_match = re.search(r'CDSs \(total\)\s+::\s+([\d,]+)', content)
cds = cds_match.group(1).replace(',', '') if cds_match else 'N/A'
# Extract tRNAs
trna_match = re.search(r'tRNAs\s+::\s+([\d,]+)', content)
trna = trna_match.group(1).replace(',', '') if trna_match else 'N/A'
# Extract rRNAs (e.g., "1, 1, 1 (5S, 16S, 23S)" -> count the numbers)
rrna_match = re.search(r'rRNAs\s+::\s+([\d,\s]+)', content)
if rrna_match:
rrna_str = rrna_match.group(1).strip()
rrna_count = sum(1 for x in rrna_str.split(',') if x.strip().isdigit())
rrna = str(rrna_count)
else:
rrna = 'N/A'
return coverage, genes, cds, trna, rrna
# 2. Parse seqkit stats
if os.path.exists("assembly_stats.tsv"):
with open("assembly_stats.tsv", "r") as f:
lines = f.readlines()
for line in lines[1:]:
parts = line.strip().split("\t")
fname = parts[0]
# Find which strain this file belongs to
target_strain = None
for strain, paths in files.items():
if paths["fna"] in fname or fname.endswith(paths["fna"]):
target_strain = strain
break
if target_strain:
idx = list(files.keys()).index(target_strain)
data["Genome size (bp)"][idx] = parts[4] # sum_len
data["Contig count"][idx] = parts[3] # num_seqs
data["Contigs N50 (bp)"][idx] = parts[12] # N50
data["Contigs L50"][idx] = parts[13] # L50
data["Guanine-cytosine content (%)"][idx] = parts[14].replace("%", "")
# 3. Parse CheckM2
if os.path.exists("checkm2_out/quality_report.tsv"):
with open("checkm2_out/quality_report.tsv", "r") as f:
lines = f.readlines()
header = lines[0].strip().split("\t")
comp_idx = header.index("Completeness")
cont_idx = header.index("Contamination")
for line in lines[1:]:
parts = line.strip().split("\t")
fname = parts[0]
target_strain = None
for strain, paths in files.items():
if paths["fna"] in fname or fname.endswith(paths["fna"]):
target_strain = strain
break
if target_strain:
idx = list(files.keys()).index(target_strain)
data["Completeness (%)"][idx] = parts[comp_idx]
data["Contamination (%)"][idx] = parts[cont_idx]
# 4. Parse GenBank files & Estimate Reads
READ_LENGTH = 150 # Change to 250 if you used Illumina 2x250bp sequencing
for strain, paths in files.items():
idx = list(files.keys()).index(strain)
gb_data = parse_gb_file(paths["gb"])
if gb_data:
coverage, genes, cds, trna, rrna = gb_data
data["Coverage depth (sequencing depth)"][idx] = f"{coverage}x"
data["Number of genes"][idx] = genes
data["Number of coding sequences (CDSs)"][idx] = cds
data["Number of tRNAs"][idx] = trna
data["Number of rRNAs"][idx] = rrna
# Estimate Total Reads: (Coverage * Genome Size) / (2 * Read Length)
try:
cov_val = float(coverage)
genome_size = int(data["Genome size (bp)"][idx].replace(',', ''))
estimated_reads = int((cov_val * genome_size) / (2 * READ_LENGTH))
data["Total number of reads sequenced"][idx] = f"~{estimated_reads:,}"
except (ValueError, TypeError):
data["Total number of reads sequenced"][idx] = "N/A (Check FASTQ)"
# 5. Output the table
df = pd.DataFrame(data)
print(df.to_markdown(index=False))
# Save to TSV for easy copy-pasting into Word/Excel
df.to_csv("final_genome_table.tsv", sep="\t", index=False)
print("\n✅ Table successfully saved to final_genome_table.tsv")Run the script:
python3 generate_table_from_gb.pyN/A or add a footnote to your manuscript table stating: “Calculated via BV-BRC EvalCon tool; not available via local CLI.” (As noted previously, these are proprietary BV-BRC machine-learning metrics).~X,XXX,XXX) based on the PGAP coverage and genome size. If your journal requires the exact raw read count, you can get it by running: zcat your_raw_R1.fastq.gz | echo $(( $(wc -l) / 4 )) and adding R1 + R2 together. However, the estimated value is usually perfectly acceptable for “Summary of sequence data” tables when derived from the assembler’s reported coverage.COMMENT block guarantees the numbers in your table perfectly match the metadata of the files you are submitting to NCBI.You are absolutely right to be cautious. The “Genome Coverage” reported in the PGAP GenBank comment is often an estimate provided by the submitter or the assembler, and it may not reflect the exact, post-mapping average depth.
Here is how you can calculate the exact coverage from your mapping files, followed by the crucial truth about calculating Coarse and Fine Consistency.
The most accurate way to calculate coverage is from the mapping file (BAM), not the raw FASTQ. If you aligned your raw reads to your assembled .fna files, you already have (or can easily generate) a BAM file.
We will use mosdepth, which is the modern, ultra-fast standard for calculating sequencing depth.
# 1. Install mosdepth (if not already installed)
conda install -c bioconda mosdepth -y
# 2. Calculate coverage for each genome
# Assuming you have a BAM file for each assembly (e.g., A6WT_mapped.bam)
# Replace 'genome.fna' and 'mapped.bam' with your actual filenames
for bam in *.bam; do
prefix=$(basename "$bam" .bam)
echo "Calculating coverage for $prefix..."
# -n means no per-base depth (much faster), -t is threads
mosdepth -n -t 4 "${prefix}_cov" genome.fna "$bam"
# Extract the mean coverage from the global distribution file
# The file looks like: length bases mean min max
mean_cov=$(awk 'NR==2 {print $3}' "${prefix}_cov.global.dist.txt")
echo "Mean Coverage for $prefix: ${mean_cov}x"
done(Note: If you don’t have BAM files yet, you can generate them quickly using minimap2 and samtools: minimap2 -ax sr genome.fna reads_R1.fq.gz reads_R2.fq.gz | samtools sort -o mapped.bam)
If you do not have BAM files and only have the raw FASTQ files, you can calculate a highly accurate estimate based on total sequenced bases divided by genome size.
# Run this in the directory containing your .fna and .fastq.gz files
# Adjust READ_LENGTH if your sequencing was 2x250bp instead of 2x150bp
READ_LENGTH=150
for fna in *.fna; do
prefix=$(basename "$fna" .fna)
# Get genome size from seqkit
genome_size=$(seqkit stats "$fna" | awk 'NR==2 {print $5}')
# Find corresponding FASTQ files (ADJUST THESE PATTERNS to match your actual file names!)
# Example: A6WT_R1.fastq.gz and A6WT_R2.fastq.gz
R1="${prefix}_R1.fastq.gz"
R2="${prefix}_R2.fastq.gz"
if [[ -f "$R1" && -f "$R2" ]]; then
# Count total lines in both files, divide by 4 to get total read pairs, multiply by 2 for total reads
total_reads=$(zcat "$R1" "$R2" | wc -l)
total_reads=$(( total_reads / 4 ))
# Calculate coverage: (Total Reads * Read Length) / Genome Size
coverage=$(echo "scale=2; ($total_reads * $READ_LENGTH) / $genome_size" | bc)
echo "$prefix: Estimated Coverage = ${coverage}x (Genome size: $genome_size bp, Total reads: $total_reads)"
else
echo "Warning: Could not find FASTQ files for $prefix. Please adjust the R1/R2 naming pattern in the script."
fi
doneYou cannot calculate “Coarse Consistency” and “Fine Consistency” using standard local Ubuntu command-line tools.
As stated in your manuscript footnote, these are proprietary metrics generated exclusively by the BV-BRC (Bacterial and Viral Bioinformatics Resource Center) platform using their EvalCon tool. This tool relies on a proprietary, machine-learning-derived catalog of ~1,300 functional roles with predictable relationships that is hosted on their servers. There is no open-source, standalone CLI equivalent (like CheckM or BUSCO) that outputs these exact two named metrics.
Option 1: The BV-BRC Web Interface (Easiest & Recommended)
.fna files.Option 2: BV-BRC Command Line Interface (If you absolutely must automate it) BV-BRC does have a CLI, but it requires setting up an API key.
# 1. Install the BV-BRC CLI (requires Node.js)
npm install -g @bvr/cli
# 2. Login to get your token (you will be prompted for your BV-BRC credentials)
bvr login
# 3. Submit a genome evaluation job (example for one file)
# This submits the job to their cloud servers
bvr submit genome-evaluation --input A6WT_chr_plasmids.fna --output eval_A6WT
# 4. Check job status and download results
bvr job-status
<JOB_ID>
bvr download
<JOB_ID> --output eval_A6WT_results.tsv
# 5. Extract the specific columns from the downloaded TSV
awk -F'\t' 'NR==1 {for(i=1;i<=NF;i++) if($i=="Coarse Consistency" || $i=="Fine Consistency") print i}' eval_A6WT_results.tsv(Note: Option 1 is vastly simpler for just 4 genomes).
If your co-authors or the journal decide that relying on a web platform for two metrics is inconvenient, the universally accepted local command-line alternative for assessing genome annotation consistency and completeness is BUSCO (Benchmarking Universal Single-Copy Orthologs).
While it doesn’t use the terms “coarse/fine consistency”, it provides “Complete (Single/Copy)”, “Fragmented”, and “Missing” percentages, which reviewers universally accept as the gold standard for genome quality.
# 1. Install BUSCO
conda install -c bioconda busco -y
# 2. Run BUSCO on your assemblies (using the bacteria_odb10 lineage)
# -m geno means genome mode, -c is threads
for fna in *.fna; do
prefix=$(basename "$fna" .fna)
busco -i "$fna" -o busco_${prefix} -l bacteria_odb10 -m genome -c 4 --auto-lineage-euk
done
# 3. View the short summary which contains the consistency/completeness metrics
cat busco_A6WT_chr_plasmids/short_summary.specific.bacteria_odb10.A6WT_chr_plasmids.txtmosdepth (Part 1, Option A) to get the exact, defensible Coverage depth.For 16S rRNA amplicon sequencing data, you should submit your raw data to the Sequence Read Archive (SRA), organized under a BioProject and linked to individual BioSamples.
You should not submit raw 16S sequencing data primarily to GEO (Gene Expression Omnibus).
Here is a breakdown of why, how the NCBI submission hierarchy works, and how to correctly link your data if processed files are also required by the journal.
When you submit, you will build the submission in this exact order:
BioProject (The Umbrella)
BioSample (The Biological Source)
SRA (The Sequencing Data)
If the target journal require you to also submit processed data (e.g., the final OTU/ASV abundance table used to generate Figures 4 and 5) to GEO, NCBI provides a streamlined workflow to avoid duplicate uploads.
GEO submissions require a metadata spreadsheet. Metadata refers to descriptive information about the overall study, individual samples, all protocols, and references to processed and raw data file names. Information is supplied by completing all fields of a metadata template spreadsheet (guidelines are provided within the file).
💡 Important: Provide enough details so that users can get a general understanding of the study and samples from the GEO records. Please spell out all acronyms and abbreviations. Submit a separate metadata spreadsheet for each data type.
If you already have your raw data in SRA, you do not need to submit it again to GEO. NCBI only needs the processed data and a specialized metadata file in order to create GEO records and link them to your raw data records previously submitted to SRA.
To do this, you must choose the second option in the GEO submission portal: 👉 “Download metadata spreadsheet with SRA accessions”
sample_name, bioSample_model (mouse), sex, age, tissue (feces), collection_date, and treatment (e.g., “post-stroke day 3”, “pre-FMT baseline”).Pro Tip: If the journal is flexible, processed data can often just be included as a Supplementary File (e.g., a .csv or .xlsx file) with the manuscript, or deposited in a repository like Figshare or Zenodo. However, if GEO is explicitly requested, the two-step SRA-first workflow above is the correct and most efficient method.
TODOs: drafting the BioProject abstract and formatting the BioSample metadata spreadsheet!
I am organizing the 16S rRNA sequencing data for deposition in the NCBI Sequence Read Archive (SRA).
Because only a subset of the sequenced samples was ultimately used in the final manuscript figures, I have compiled an exact list of the specific samples to be submitted. I want to ensure our public dataset perfectly aligns with the content of the manuscript.
Here is the exact list of samples I will upload to NCBI, mapped to the manuscript figures. Could you please confirm that they are correct?
1. Stroke Model Groups (Used in Fig. 4D–F for blood/brain SCFA)
2. Baseline Donor Groups (Used in Fig. 4A–C, Supp. Fig. 4, Fig. 5C)
3. Pre-FMT Baseline Groups (Used in Fig. 5B as purple dots, n=18 total)
4. FMT Recipient Groups (Used in Fig. 5B, 5C, 5D, 5E)
Points for Your Final Confirmation:
这个陷阱在纪录片中得到了解决方法。
很多人以为贫穷是因为赚得少,但这四个人里,泰兹赚得并不少,金夫妇的收入也过了中位数。
他们贫穷的根源在于情绪消费。
安娜用购物填补童年的空洞,琳赛用美食犒劳辛苦的自己,金夫妇用昂贵的玩具表达对孩子的爱,弥补陪伴的不足。
在消费主义盛行的时代,商家最擅长的就是把「商品」和「幸福」划等号。我们买的不是东西,而是那一瞬间的多巴胺。
然而,多巴胺消退后,留下的只有账单和空虚。
另一个看不见的敌人是通胀。专家告诉泰兹,把钱单纯存在银行里是不安全的,专家建议他将一部分积蓄定期投入标普500指数基金。
泰兹听从了建议,他的资产不仅保全了,还实现了大幅增值。反之,如果他仅仅持有现金,购买力已经被通胀侵蚀了大半。
金夫妇为了表达爱意,花了很多钱给孩子买玩具,但孩子在这样的环境中长大,会自然而然地认为「钱是用来花的」「想要就必须马上得到」。财务观念的缺失,比一时贫穷更可怕。
即便「提前退休」似乎很难实现,但他们采用了「迷你退休」。一年中某一段时间,推掉所有工作,一家人在一起度假,孩子们也不再需要更多玩具。
在理财专家介入后,这四组人的生活发生了变化。
琳赛辞去了那份消耗她精力的服务工作,她开始一边摆地摊卖宠物主题的画作,一边帮人遛狗。虽然收入不稳定,但她有了更多时间创作。「做梦钱」账户数字的增加,给了琳赛更多安全感,她甚至有余力去做心理咨询,处理内心的焦虑。
泰兹开始正视自己的财务状况。他严格控制开支,系统性地学习理财知识。虽然因为伤病无法重返巅峰,但他利用运动员时期的积蓄进行了稳健投资。
亚莉安娜把债务进行排序了:先还利率最高的卡,再往下滚,不让利息吃掉还款进度。一年后,她的卡债逐渐被有序还清,开始有里应急金和一点存款。
金夫妇开始记录自己的每一笔开销。他们惊讶地发现,仅仅是减少外出就餐和停止购买不必要的玩具,每个月就能省下2000美元。
他们不再是金钱的奴隶,而是成为了自己生活的主人。
结语
前段时间我去越南旅行。
因为电子支付不普及,我换了大量的越南盾现金。当地最大面额是50万盾,约合人民币128元。
我体验到了久违的「花钱如流水」的感觉。手里厚厚的钞票变薄,「肉疼」的感觉非常直观。即便越南物价低廉,我依然觉得钱花得太快了。
一回到国内,打开手机里的支付宝和微信,那种痛感消失了。短短一周,我在网上的消费已经远超在越南的开支。
我意识到,电子支付剥离了「钱」的物理属性,让消费变成数字,极大地降低了我们的痛感阈值。
基于此,我想给大家几点具体的建议:
先看水流,再看水源
如果你的池子到处是漏洞,注入再多水也会流干。先花一个月时间记账,看清钱的流向,你会发现至少30%的开销是不必要的。
建立物理隔离
发工资的第一时间,先划出“储蓄”和“投资”的部分,剩下的才是生活费。千万不要把所有钱放在一个活期账户里任由支配。
警惕小额高频消费
每天一杯35元的咖啡,一年就是一万多;路过便利店买个零食,一年又是几千。养成记账的习惯,只有看见,才能改变。
财务自由并不只是关于钱,它是关于选择,关于自由,关于能否掌控自己的生活。那些被我们忽视的——开源、节流、投资——就是生活的本身。
| 特色功能 | AirPods 4 (一般款) | AirPods 4 (主動降噪款) | AirPods Pro 2 |
|---|---|---|---|
| 推出時間 | 2024年9月 | 2024年9月 | 2022年9月 |
| 佩戴設計 | 半開放式,無耳塞 | 半開放式,無耳塞 | 入耳式,附矽膠耳塞 |
| 晶片 | H2 晶片 | H2 晶片 | H2 晶片 |
| 主動降噪 (ANC) | 無 | 有(效果約為Pro 2的一半) | 有(頂級降噪效果) |
| 通透模式 | 無 | 有 | 有 |
| 適應式音訊 | 無 | 有 | 有 |
| 對話感知 | 無 | 有 | 有 |
| 聽力健康功能 | 無 | 無 | 有(聽力測試、助聽器功能、降低高音量) |
| 單次續航 (ANC開啟) | 最長5小時 | 最長4小時 | 最長6小時 |
| 搭配充電盒總續航 | 最長30小時 | 最長30小時 | 超過24小時 |
| 充電盒功能 | USB-C充電,無無線充電 | USB-C充電,支援無線充電,內建揚聲器支援尋找功能 | MagSafe充電盒(USB-C),支援無線充電,內建揚聲器與U1晶片支援精確尋找,有掛繩孔 |
| 抗汗抗水 | IP54 (防塵抗水) | IP54 (防塵抗水) | IP54 (防塵抗水) |
在目前三款產品中,只有 AirPods Pro 2 具備完整的聽力健康功能**。
具體包含以下三個面向:
答案是:AirPods 4 是最新款。
簡單來說:
脚本:MicrobiotaProcess_PCA_Group3-4.R
对应图表:论文 图 4A–C
| 图号 | 内容 | 分析方法 |
|---|---|---|
| 4A | 实验设计示意图 | – |
| 4B | PCoA 图(Bray-Curtis 距离) | mp_cal_pcoa() + mp_plot_ord() |
| 4C | 差异丰度 OTU 气泡图 | DESeq2 + 气泡图可视化 |
该分析比较的是老年雄性 vs 老年雌性小鼠的稳态肠道微生物组成
脚本:MicrobiotaProcess_Group9_10_11_PreFMT.R + Phyloseq_Group9_10_11_pre-FMT.Rmd
对应图表:论文 图 5B、5C、5D、5E
| 图号 | 内容 | 分析方法 |
|---|---|---|
| 5B | FMT 后受体小鼠的 PCoA 图 | mp_cal_pcoa() + mp_plot_ord() |
| 5C | 科水平相对丰度堆叠图 | mp_plot_abundance(taxa.class = Class) |
| 5D | 差异丰度 OTU 气泡图(FMT 组间比较) | DESeq2/LEfSe + 气泡图 |
| 5E | 脑内 IL-17A⁺ γδ T 细胞流式定量 | 流式细胞术 + 统计检验 |
该分析验证粪便微生物移植(FMT)对年轻受体小鼠卒中后免疫反应的影响
mp_adonis) + 事后两两比较mp_plot_ord 用于序贯图,mp_plot_abundance 用于组成图以下是本研究中使用的全部 14 个实验组别的详细说明,按功能分类整理:
| 组号 | 样本前缀 | 完整样本 | 性别/年龄 | 状态 | 用途 |
|---|---|---|---|---|---|
| 1 | sample-A* |
A1–A11 | ♂ 老年 | 中风后 3 天 | 图 4D–F(血液/脑组织 SCFA 检测) |
| 2 | sample-B* |
B1–B16 | ♀ 老年 | 中风后 3 天 | 图 4D–F(血液/脑组织 SCFA 检测) |
📌 说明:这两组用于比较中风后老年雄性和雌性小鼠的微生物代谢物(短链脂肪酸)水平差异。
| 组号 | 样本前缀 | 完整样本 | 性别/年龄 | 状态 | 用途 |
|---|---|---|---|---|---|
| 3 | sample-C* |
C1–C10 | ♀ 老年 | 基线,FMT 供体 | 图 4A–C(16S 测序)、补充图 4、图 5C(Boxplot 3) |
| 4 | sample-E* |
E1–E10 | ♂ 老年 | 基线,FMT 供体 | 图 4A–C(16S 测序)、补充图 4、图 5C(Boxplot 2) |
| 5 | sample-F* |
F1–F5 | ♂ 年轻 | 基线,FMT 供体 | 对照供体,未在主图中展示 |
📌 关键说明:
- 组 3 和组 4 是粪菌移植(FMT)的供体小鼠,用于提供老年雌/雄肠道菌群
- 图 4 和补充图 4 中实际使用的样本为:♀供体 C1–C6(n=6),♂供体 E1–E8(n=8),其余样本因年龄偏小或测序深度不足被排除
| 组号 | 样本前缀 | 完整样本 | 性别/年龄 | 状态 | 用途 |
|---|---|---|---|---|---|
| 6 | sample-G* |
G1–G6 | ♂ 老年 | FMT 前,抗生素处理前,批次 I | 图 5B(紫色,pre-FMT 基线) |
| 7 | sample-H* |
H1–H6 | ♀ 老年 | FMT 前,抗生素处理前,批次 I | 图 5B(紫色,pre-FMT 基线) |
| 8 | sample-I* |
I1–I6 | ♂ 年轻 | FMT 前,抗生素处理前,批次 II | 图 5B(紫色,pre-FMT 基线) |
📌 说明:这三组合并为”pre-FMT”基线组(n=18),代表年轻雄性受体小鼠在接受粪菌移植之前的肠道菌群状态。
| 组号 | 样本前缀 | 完整样本 | 性别/年龄 | 接受供体 | 状态 | 用途 |
|---|---|---|---|---|---|---|
| 9 | sample-J* |
J1–J4, J10, J11 | ♂ 年轻 | 老年♂供体 | FMT 后,中风前 | 图 5B🔵、5C(Boxplot 4)、5D、5E |
| 10 | sample-K* |
K1–K6 | ♂ 年轻 | 老年♀供体 | FMT 后,中风前 | 图 5B🔴、5C(Boxplot 5)、5D、5E |
| 11 | sample-L* |
L2–L6 | ♂ 年轻 | 年轻♂供体 | FMT 后,中风前 | 图 5B🟢、5E(对照) |
📌 关键说明:
- 所有受体均为年轻雄性小鼠,仅供体来源不同
- “aged♂ FMT” = 接受老年雄性供体粪便的年轻受体(不是受体本身是老年!)
- 图 5C 的 5 个箱线图 = pre-FMT 基线 + 2 个供体组 + 2 个受体组(不含年轻♂供体受体组)
| 组号 | 样本前缀 | 完整样本 | 性别/年龄 | 接受供体 | 状态 | 用途 |
|---|---|---|---|---|---|---|
| 12 | sample-M* |
M1–M8 | ♂ 老年 | 老年♂供体 | FMT 后,中风后 | 补充分析 |
| 13 | sample-N* |
N1–N10 | ♀ 老年 | 老年♀供体 | FMT 后,中风后 | 补充分析 |
| 14 | sample-O* |
O1–O8 | ♂ 年轻 | 年轻♂供体 | FMT 后,中风后 | 补充分析 |
📌 说明:这三组用于探索性分析,未出现在主论文图表中。
✅ "FMT 标签 = 供体特征,不是受体特征"
• aged♂ FMT = 供体是老年雄性
• 受体永远是年轻雄性(本实验设计)
✅ 图 4 = 老年供体(组 3/4)+ 老年中风小鼠(组 1/2)
✅ 图 5 = FMT 实验:受体(组 6–11)+ 供体(组 3/4)
✅ 补充图 4 = 仅老年供体(组 3/4,筛选后 C1–C6, E1–E8)| 组别 | 排除样本 | 排除原因 |
|---|---|---|
| 组 3(♀供体) | C7, C8, C9, C10 | C8–C9 年龄偏小;C10 为离群值/测序深度低 |
| 组 4(♂供体) | E9, E10 | 测序深度低/离群值 |
| 组 9(受体) | J5, J6, J7, J8, J9 | 测序深度不足或质量控制排除 |
| 组 10(受体) | K7–K15 | 测序深度不足或质量控制排除 |
| 组 11(受体) | L1, L7–L15 | 测序深度不足或质量控制排除 |
📌 最终用于分析的样本数以各图图例标注为准(如:图 5 中 aged♂ FMT n=6, aged♀ FMT n=6)
TODO: 导出完整的样本–组别映射 CSV 文件,or 提供某张图的精确样本列表🎯
aged♂ FMT = 接受了老年雄性供体粪便的年轻雄性受体小鼠
| 角色 | 年龄/性别 | 说明 |
|---|---|---|
| 受体(接受粪便) | 🐭 年轻雄性(4周龄起始) | 所有 FMT 组的受体都是相同的年轻雄性小鼠 |
| 供体(提供粪便) | 🐭 老年雄性 / 老年雌性 / 年轻雄性 | 供体的年龄/性别是实验变量 |
🟣 Purple (pre-FMT, n=18): G1–G6, H1–H6, I1–I6
→ FMT前的基线年轻雄性小鼠(未接受移植)
🔵 Blue (aged♂ FMT, n=6): J1, J2, J3, J4, J10, J11
→ 年轻雄性受体 + 接受【老年雄性】供体粪便
🔴 Red (aged♀ FMT, n=6): K1–K6
→ 年轻雄性受体 + 接受【老年雌性】供体粪便
🟢 Green (young♂ FMT, n=5): L2–L6
→ 年轻雄性受体 + 接受【年轻雄性】供体粪便(对照组)来自 260311_LTPaper.pdf Figure 5 图例:
“Principal coordinates analysis (PCoA) of young male mice before (purple) (n=18), and after FMT of aged male (n=6) (blue) or female (n=6) (red) or young male (n=5) (green) stool donors.”
→ 明确说明分析对象是 young male mice,括号内描述的是 stool donors(粪便供体)的特征。
来自 Supplemental Methods “Microbiota eradication and FMT”:
“4 weeks old male mice were treated for 2 weeks with an antibiotic cocktail… recipient mice were gavaged with donor stool four times over two weeks.”
→ 受体小鼠起始年龄为 4周龄(年轻)。
Figure 5 小标题:
“FMT of aged male microbiota increases IL-17A-producing γδ T cells in the post-ischemic brain of young recipient mice“
→ 再次确认受体是 young recipient mice。
这个实验的核心科学问题是:
“供体微生物的年龄/性别特征,能否通过移植’传递’给受体,并影响受体的免疫反应?”
通过保持受体一致(年轻雄性),仅改变供体来源,可以:
“FMT 标签 = 供体特征,不是受体特征”
- aged♂ FMT = 供体是老年雄性
- 受体永远是年轻雄性(本实验中)
“Principal coordinates analysis (PCoA) of young male mice before (purple) (n=18), and after FMT of aged male (n=6) (blue) or female (n=6) (red) or young male (n=5) (green) stool donors.”
G1–G6, H1–H6, I1–I6J1, J2, J3, J4, J10, J11K1–K6L2–L6 (L1, L7–L15 excluded for low depth/QC)Based on your co-author’s note: “Figure 5C uses the Figure 5B recipient samples PLUS the aged donor samples (Groups 3 & 4).”
G1–G6, H1–H6, I1–I6E1–E10 C1–C7 J1, J2, J3, J4, J10, J11K1–K6⚠️ Key difference: Group11 (young♂ FMT recipients,
L2–L6) is shown in Figure 5B but is NOT included in Figure 5C, since Figure 5C focuses on comparing the effect of aged donor microbiota.
“Bubble plot showing differentially abundant Operational Taxonomic Units (OTUs) between young male recipients of aged female vs. aged male FMT. x-axis = log₂ fold change, y-axis = bacterial family, bubble size = adjusted p-value, color = bacterial order.”
J1, J2, J3, J4, J10, J11 → Reference group (log₂FC < 0 = enriched in this group)K1–K6 → Comparison group (log₂FC > 0 = enriched in this group)| Key families highlighted in the plot: | Direction | Family (Order) | Enriched in | Biological note |
|---|---|---|---|---|
| 🔴 Positive log₂FC | Lachnospiraceae (Clostridiales) | Aged♀ FMT | SCFA producer | |
| 🔴 Positive log₂FC | Ruminococcaceae (Clostridiales) | Aged♀ FMT | SCFA producer | |
| 🔴 Positive log₂FC | Muribaculaceae (Bacteroidales) | Aged♀ FMT | SCFA producer | |
| 🔴 Positive log₂FC | Desulfovibrionaceae (Desulfovibrionales) | Aged♀ FMT | Sulfate-reducing | |
| 🔵 Negative log₂FC | Erysipelotrichaceae (Erysipelotrichales) | Aged♂ FMT | Pro-inflammatory association | |
| 🔵 Negative log₂FC | Rikenellaceae (Bacteroidales) | Aged♂ FMT | Context-dependent | |
| 🔵 Negative log₂FC | Clostridiales vadinBB60 group | Aged♂ FMT | Function unclear |
⚠️ Note: This analysis uses DESeq2 on non-rarefied integer counts from
ps_filt, with taxa prefiltered (total counts ≥10). Only taxa with Benjamini–Hochberg adjusted p < 0.05 are shown. The same ASVs/OTUs appear in Figure 4C and Supplementary Figure 4B, but Figure 5D specifically compares FMT recipient outcomes (Groups 9 vs. 10), not baseline donor differences.
“(A) Bray-Curtis distances between aged male-male, female-female and female-male stool samples under homeostatic conditions (nmale=8 and nfemale=6). (B) Cladogram showing differentially abundant OTUs…”
E1–E8 (E9, E10 excluded for low sequencing depth/outliers)C1–C6 (C7–C10 excluded; C8–C9 younger mice, C10 outlier)“We profiled the gut bacterial composition of aged male and female mice by 16S rRNA-seq…”
C1–C6 E1–E8 (Note: Figure 4D–F show SCFA concentrations measured by targeted UHPLC-MS/MS, not 16S data.)
Your observation is CORRECT: PICRUSt2 results are NOT used in Figure 4D–F.
| Question | Answer | Evidence |
|---|---|---|
| Are PICRUSt2 results used in Figure 4? | ❌ No | Figure 4D–F legend explicitly states: “measured by targeted mass spectrometry” |
| Are PICRUSt2 results used anywhere in the manuscript? | ❌ No evidence | README_PICRUSt2.txt files contain exploratory pipeline notes, but no PICRUSt2 figures, tables, or text appear in 260311_LTPaper.pdf or 260310_Supplements.pdf |
| Is the SCFA data in Figure 4D–F experimentally measured? | ✅ Yes | Supplemental Methods (pages 12–13) describe UHPLC-MS/MS quantification with internal standards, derivatization, and MRM parameters |
Key distinction:
Here is the merged quick reference table combining Figure 5B, 5C, and 5D with related figures, formatted for easy copy-paste:
| Figure | Comparison | Sample IDs (exact) | n | Purpose |
|---|---|---|---|---|
| Figure 4B-C | Aged♀ vs. aged♂ donors (homeostatic) | C1–C6 vs. E1–E8 |
6 vs. 8 | Baseline sex differences in microbiota (DESeq2 bubble plot) |
| Suppl Fig 4B | Same as Fig 4C | C1–C6 vs. E1–E8 |
6 vs. 8 | Phylogenetic context of differential taxa (cladogram) |
| Figure 5B | Pre-FMT vs. post-FMT recipients (4-group PCoA) | G1–G6, H1–H6, I1–I6 (pre-FMT); J1, J2, J3, J4, J10, J11 (aged♂ FMT); K1–K6 (aged♀ FMT); L2–L6 (young♂ FMT) |
18, 6, 6, 5 | PCoA: microbiome shift after FMT (Bray–Curtis) |
| Figure 5C | Donors vs. recipients (5 boxplots, family-level) | G1–G6, H1–H6, I1–I6 (pre-FMT); E1–E8 (aged♂ donors); C1–C6 (aged♀ donors); J1, J2, J3, J4, J10, J11 (aged♂ FMT); K1–K6 (aged♀ FMT) |
18, 8, 6, 6, 6 | Taxonomic composition: donors vs. recipients (relative abundance) |
| Figure 5D | Aged♀ vs. aged♂ FMT recipients (DESeq2) | K1–K6 vs. J1, J2, J3, J4, J10, J11 |
6 vs. 6 | Effect of donor microbiota on recipient immune response (differential abundance) |
| Figure 5E | Same recipients as Fig 5D (+ young♂ control) | K1–K6 vs. J1, J2, J3, J4, J10, J11 (+ L2–L6) |
6 vs. 6 (+5) | IL-17A+ γδ T cells in brain post-FMT (flow cytometry) |
| Group # | Description | Sample Prefix | Full IDs | Used In |
|---|---|---|---|---|
| 3 | Aged female, baseline FMT donor | sample-C* |
C1–C10 (C1–C6 used in Fig 4B-C, Suppl Fig 4, Fig 5C) | Fig 4C, Suppl Fig 4, Fig 5C |
| 4 | Aged male, baseline FMT donor | sample-E* |
E1–E10 (E1–E8 used in Fig 4B-C, Suppl Fig 4, Fig 5C) | Fig 4B-C, Suppl Fig 4, Fig 5C |
| 6 | Aged male, pre-antibiotics FMT batch I | sample-G* |
G1–G6 | Fig 5B (purple), Fig 5C (Boxplot 1) |
| 7 | Aged female, pre-antibiotics FMT batch I | sample-H* |
H1–H6 | Fig 5B (purple), Fig 5C (Boxplot 1) |
| 8 | Young male, pre-antibiotics FMT batch II | sample-I* |
I1–I6 | Fig 5B (purple), Fig 5C (Boxplot 1) |
| 9 | Young male, post-FMT aged male stool | sample-J* |
J1–J4, J10, J11 (J5–J9 excluded) | Fig 5B (blue), Fig 5C (Boxplot 4), Fig 5D, Fig 5E |
| 10 | Young male, post-FMT aged female stool | sample-K* |
K1–K6 | Fig 5B (red), Fig 5C (Boxplot 5), Fig 5D, Fig 5E |
| 11 | Young male, post-FMT young male stool | sample-L* |
L2–L6 (L1, L7–L15 excluded) | Fig 5B (green), Fig 5E (not in Fig 5C/D) |
ps_filt (taxa prefiltered: total counts ≥10). Only taxa with BH-adjusted p < 0.05 are shown.L2–L6) for comparison of IL-17A+ γδ T cells.Let me know if you’d like me to:
Subject: Re: Manuscript Review (Lines 276-348) & NCBI SRA Citation
Thank you for sending the manuscript and for the opportunity to review the specified sections. I have carefully reviewed lines 276–348 covering the microbiota composition analysis and FMT experiments.
I noticed a few minor typographical inconsistencies in the taxonomic nomenclature that may warrant correction before submission:
| Line | Current Text | Suggested Correction |
|---|---|---|
| 295 | Muribaculae (order Bacteroidalis) | Muribaculaceae (order Bacteroidales) |
| 298 | Ruminococcae | Ruminococcaceae |
| 334 | Muribaculae | Muribaculaceae |
These appear to be minor spelling variations; please confirm if these align with your intended taxonomic references.
The scientific content, logic flow, and figure references (Fig. 4A–D, Fig. 5A–E) are clear and well-integrated with our analysis scripts (MicrobiotaProcess_Group3-4.R and MicrobiotaProcess_Group9_10_11_PreFMT.R).
Regarding the NCBI SRA citation:
PRJNAxxxxxx) to cite in the manuscript.Dataset: 16S rRNA-seq data are deposited in the NCBI Sequence Read Archive (SRA)
under BioProject accession number [TO BE ADDED].If you confirm, I can proceed with preparing the submission files this week so we meet your timeline.
I am flexible and ready to assist with final revisions or SRA submission as needed. Please let me know the exact submission date once confirmed, and I will prioritize accordingly.
Thank you again, and I wish you a pleasant weekend as well!
Note for me: Before sending,:
- Double-check the taxonomic spellings against your reference database (SILVA/GTDB)
- Confirm whether Marius prefers to handle the SRA upload himself or delegate it
- Attach the prepared metadata template if you want to expedite the process
Would you like me to help draft the NCBI BioProject metadata table or refine any part of this reply?
(一)mBio
Immune activation of primary human macrophages is suppressed by the coordinated action of Yersinia effectors
最新IF:6.747,近四年影响因子变动小,基本维持在6左右;中科院分区 1区,这个分区对于对于有毕业要求的投稿人,这个期刊性价比很高;
OA开放访问:是;
年文章数:509篇;
投稿周期:官方时间为平均3天左右筛选Editor,平均35天到第一个决定,接收到online平均22天,这个时间相对来说还是友好的,文章只online发布;对于赶时间,且研究方向符合此期刊的,不妨可以考虑;
接收文章类型:精简性综述和研究类,此期刊在初始提交时没有格式要求,Freestyle;
接收文章偏好性:分为以下6大主题:
Applied and Environmental Science
Clinical Science and Epidemiology
Ecological and Evolutionary Science
Host-Microbe Biology
Molecular Biology and Physiology
Therapeutics and Prevention
包括但不限于生物化学和分子生物学,遗传学和基因组学,环境科学,进化,免疫学,传染病和生理学。涵盖的主题包括细菌,病毒,寄生虫,真菌和简单的真核生物,以及所有类型的宿主 – 微生物相互作用。
(二)mSystems
Chloramphenicol stress triggers oxidative adaptation in Acinetobacter baumannii ATCC19606 devoid of RND efflux pumps AdeAB or AdeIJ (Data_Foong_RNAseq_2021_ATCC19606_Cm/)
最新IF:6.519,中科院2区,从2017年开始有影响因子,起步比较高,5.75,一年跨越到6.519,可惜的是被分成了中科院2区;
OA开放访问:是;
年文章数量:134篇,即将开放同行评审,相比mBio来说,年文章数量少了很多,在理论上接收概率是偏小;
接收文章主题分为以下几大主题:
Applied and Environmental Science
Clinical Science and Epidemiology
Ecological and Evolutionary Science
Host-Microbe Biology
Molecular Biology and Physiology
Novel Systems Biology Techniques
Synthetic Biology
Therapeutics and Prevention
与mBio略有不同
对于研究方法的偏好性:微生物组,基因组学,宏基因组学,转录组学,代谢组学,蛋白质组学,生物信息学和计算微生物学的研究交叉学科;
投稿周期:官方时间平均32个自然日会出第一个决定,时间来说也是非常的快,所以每个期刊的优势不同,按需选择,没有最好,只有更好!
(三)MICROBIOL RESOUR ANN.
Draft genome sequence of Enterobacter hormaechei HKEH-1 isolated from a diabetic patient’s blood (Data_Tam_DNAseq_2025_E.hormaechei_and_Non-antibiotic_transport_on_ATCC19606/)
Genome sequences and transcriptomic data of Acinetobacter baumannii ATCC19606 devoid of efflux pumps (Data_Foong_DNAseq_ATCC19606_Cm/)
《Microbiology Resource Announcements》(MRA,中文常译作《微生物学资源公告》)是由美国微生物学会(ASM)出版的一本纯在线、完全开放获取(Open Access)的同行评审期刊 [[1]]。
以下是该期刊的简短核心特点:
简而言之,如果您有一组高质量的微生物测序数据或新构建的菌株,希望快速、规范地向学术界“注册”并公开,MRA 是一个非常合适的发表平台。
(四)Applied and Environmental Microbiology
最新IF:4.077 近四年,影响因子处于上升趋势,中科院2区,相比mBio和mSystems, 此期刊显得更加亲民;
OA开放访问:否;
年文章数:612篇;
投稿周期:2个月;
接收率:60%(数据来源网络);
接收文章偏好性:应用微生物研究的各个方面的描述,微生物生态学的基础研究,以及关注具有实用价值的微生物主题的遗传和分子性质的研究。研究必须解决显着的微生物学原理,基本微生物过程或应用或环境微生物学的基本问题。所考虑的主题包括与食品,农业,工业,生物技术,公共卫生,植物和无脊椎动物有关的微生物学以及与微生物生态学相关的细菌,真菌,藻类,原生动物和其他简单真核生物的基本生物学特性。新的重要发现,以促进对微生物学的理解,以及其他科学家可能建立的。
(五)Microbiology Spectrum
Distinct Substrate Specificities of AdeABC and AdeIJK Shape Tolerance to Human-Targeted Drugs in Acinetobacter baumannii (Data_Tam_DNAseq_2025_E.hormaechei_and_Non-antibiotic_transport_on_ATCC19606/)
《Microbiology Spectrum》(微生物学谱)是由美国微生物学会(ASM)出版的一本同行评议国际学术期刊。该期刊创刊于2013年,专注于发表微生物学领域的基础、应用与临床研究成果,涵盖病毒学、细菌学、真菌学及环境微生物生态等多个方向
(六)Others
Validation of Small-Molecule Entry Inhibitors Targeting the Respiratory Syncytial Virus (RSV)