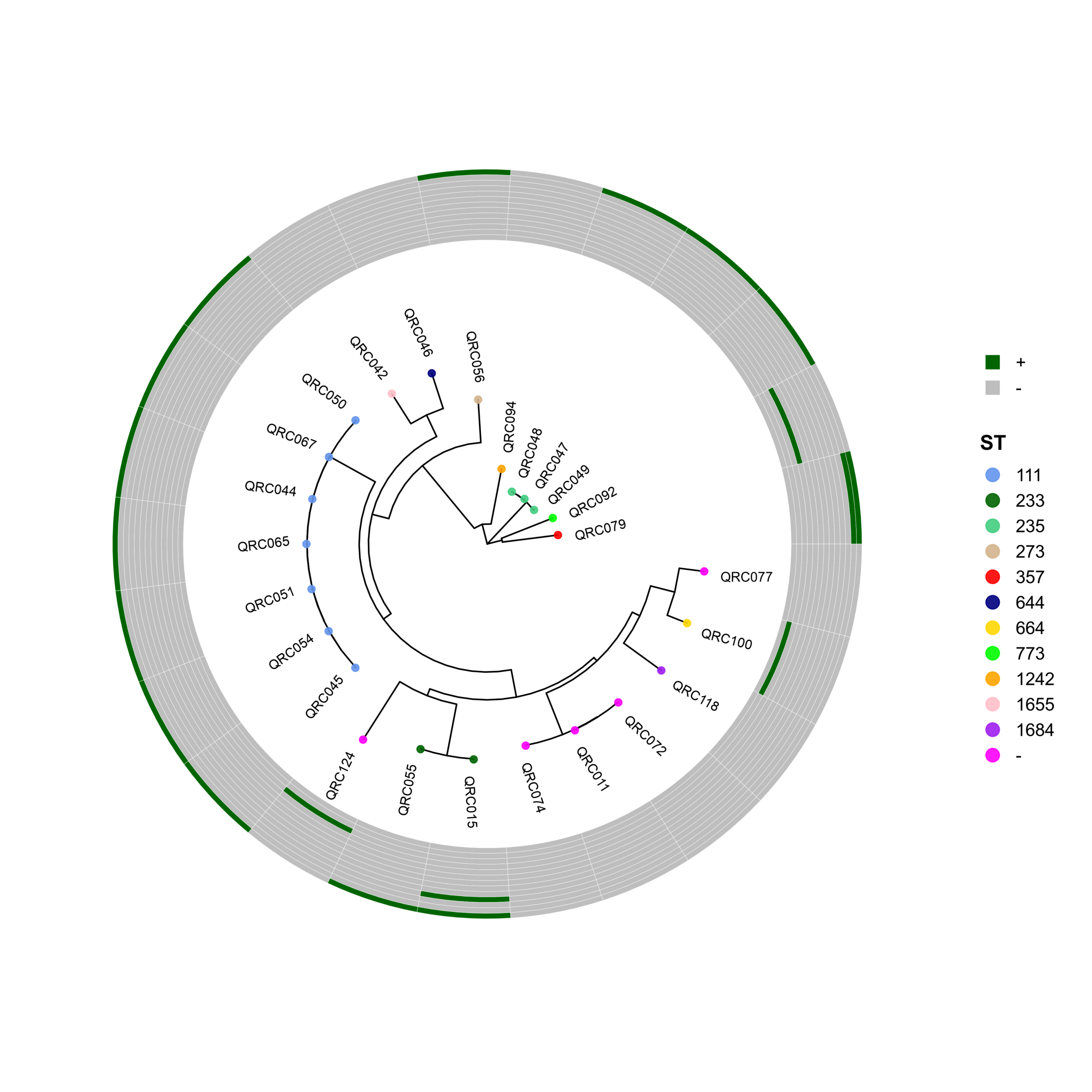
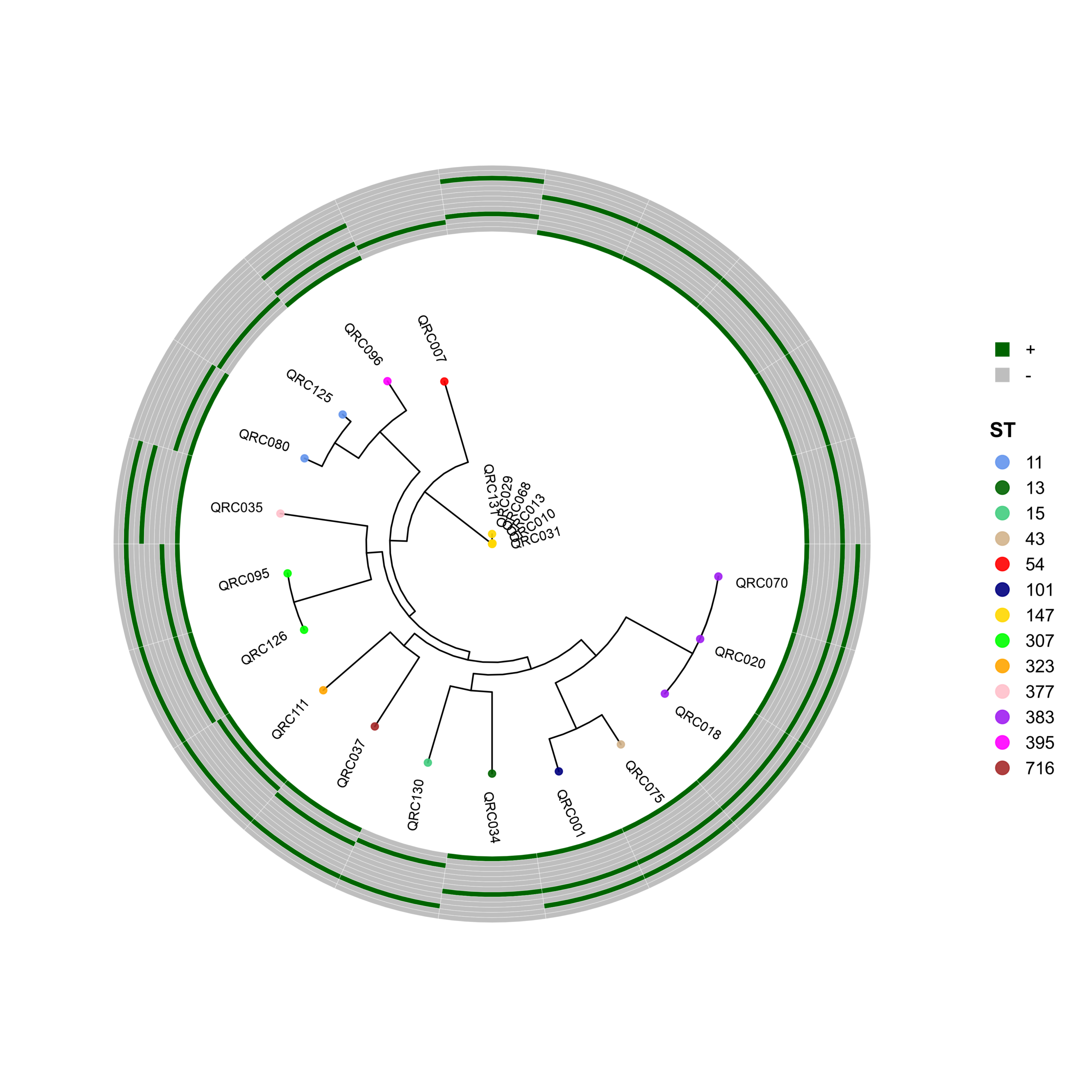
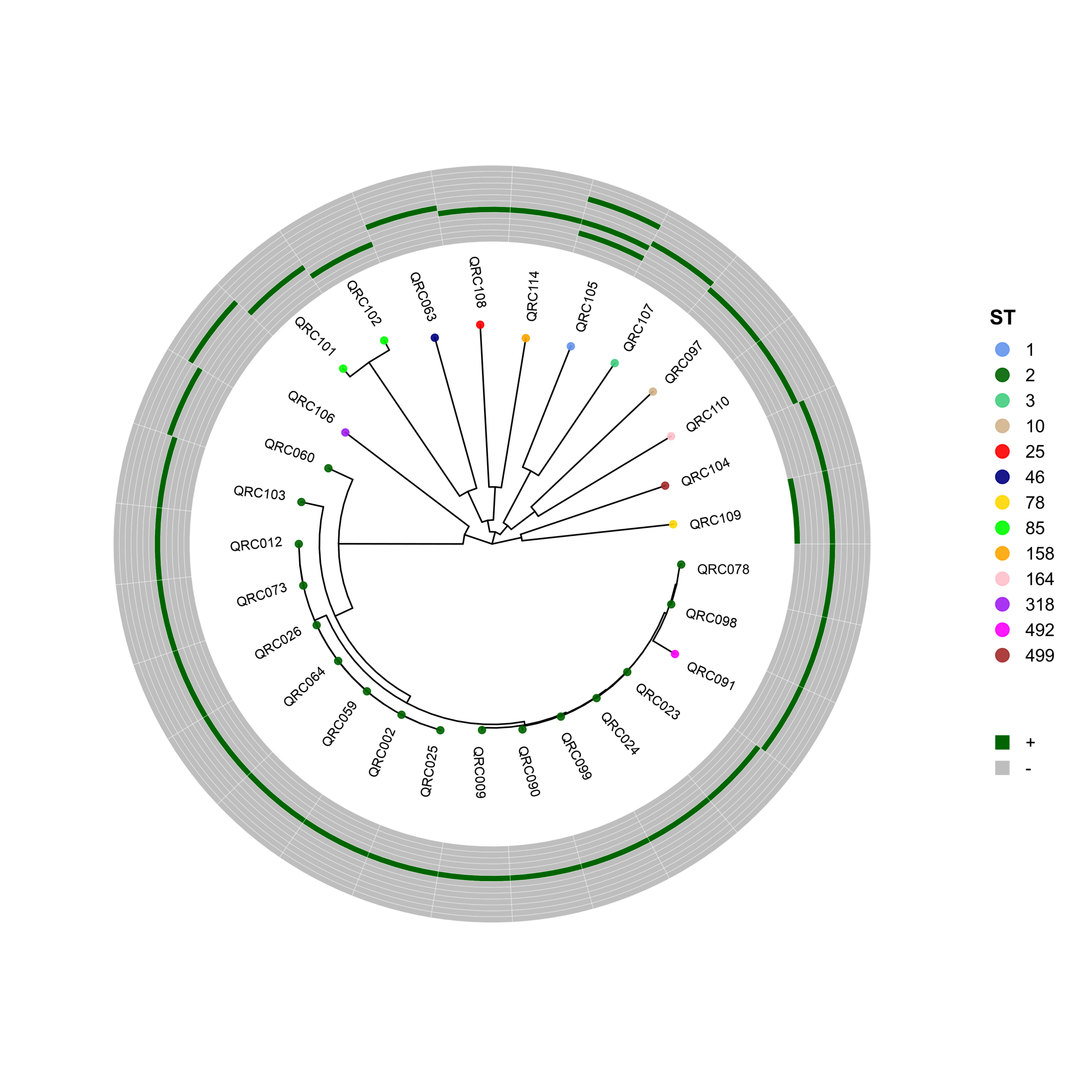
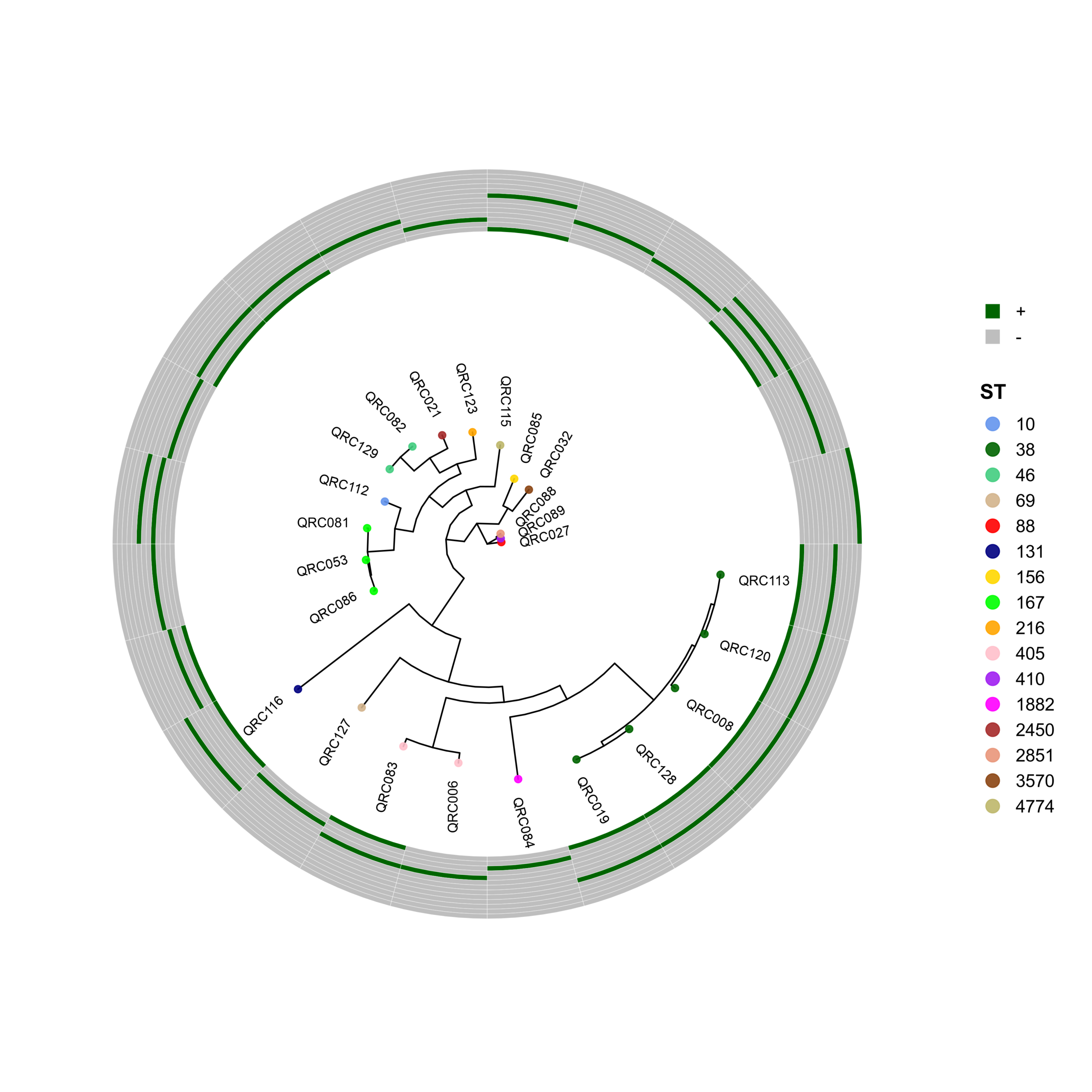
🧬 Circular Phylogenetic Tree Pipeline with Resistance Gene Heatmaps
Complete workflow for Figure S1 generation – suitable for homepage documentation
📋 Pipeline Overview
WGS Reads (100 isolates)
│
▼
┌─────────────────────┐
│ 1. Species Clustering│
│ • E. coli (n=24) │
│ • K. pneumoniae (n=22)│
│ • A. baumannii (n=30)│
│ • P. aeruginosa (n=25)│
└─────────────────────┘
│
▼
┌─────────────────────┐
│ 2. Genome Annotation│
│ • Prokka v1.14.5 │
│ • TIGRFAMs HMM DB │
│ • Output: *.gff │
└─────────────────────┘
│
▼
┌─────────────────────┐
│ 3. Pangenome Analysis│
│ • Roary v3.13.0 │
│ • Core gene alignment│
│ • Gene P/A matrix │
└─────────────────────┘
│
▼
┌─────────────────────┐
│ 4. Resistance Gene │
│ Detection │
│ • Abricate + ResFinder│
│ • 13 β-lactamase genes│
└─────────────────────┘
│
▼
┌─────────────────────┐
│ 5. Phylogenetic Tree│
│ • RAxML-NG │
│ • GTR+G model │
│ • 1000 bootstraps │
└─────────────────────┘
│
▼
┌─────────────────────┐
│ 6. Visualization │
│ • ggtree (R) │
│ • Circular layout │
│ • ST-colored tips │
│ • Gene heatmap ring │
└─────────────────────┘
🔧 Step-by-Step Commands
1️⃣ Prokka Annotation (per species)
#!/bin/bash
# prokka_run.sh
SPECIES_CONFIG=(
"ecoli:Escherichia:coli"
"kpneumoniae:Klebsiella:pneumoniae"
"abaumannii:Acinetobacter:baumannii"
"paeruginosa:Pseudomonas:aeruginosa"
)
for config in "${SPECIES_CONFIG[@]}"; do
IFS=':' read -r SPECIES_KEY GENUS SPECIES_NAME <<< "$config"
for SAMPLE in $(cat samples_${SPECIES_KEY}.txt); do
prokka --force \
--outdir prokka/${SPECIES_KEY}/${SAMPLE} \
--cpus 8 \
--kingdom Bacteria \
--genus "${GENUS}" \
--species "${SPECIES_NAME}" \
--addgenes --addmrna \
--prefix "${SAMPLE}" \
--locustag "${SAMPLE}" \
--hmm /media/jhuang/Titisee/GAMOLA2/TIGRfam_db/TIGRFAMs_15.0_HMM.LIB \
assemblies/${SAMPLE}.fasta
done
done
2️⃣ Roary Pangenome Analysis
#!/bin/bash
# roary_run.sh
# A. baumannii
roary -f roary/abaumannii -e --mafft -p 40 \
prokka/abaumannii/*/*.gff
# E. coli
roary -f roary/ecoli -e --mafft -p 40 \
prokka/ecoli/*/*.gff
# K. pneumoniae
roary -f roary/kpneumoniae -e --mafft -p 40 \
prokka/kpneumoniae/*/*.gff
# P. aeruginosa
roary -f roary/paeruginosa -e --mafft -p 40 \
prokka/paeruginosa/*/*.gff
3️⃣ Resistance Gene Detection (Abricate + ResFinder)
#!/bin/bash
# abricate_run.sh
# Setup databases (one-time)
abricate --setupdb
# Define target genes
TARGET_GENES="blaCTX-M,blaIMP,blaKPC,blaNDM-1,blaNDM-5,blaOXA-23-like,blaOXA-24-like,blaOXA-48-like,blaOXA-58-like,blaPER-1,blaSHV,blaVEB-1,blaVIM"
for SPECIES in ecoli kpneumoniae abaumannii paeruginosa; do
for SAMPLE in $(cat samples_${SPECIES}.txt); do
# Run against ResFinder
abricate --db resfinder \
--minid 90 --mincov 80 \
assemblies/${SAMPLE}.fasta \
> abricate/${SPECIES}/${SAMPLE}.resfinder.tsv
# Extract target genes to CSV-ready format
awk -F'\t' -v sample="${SAMPLE}" '
NR==1 {next}
$10 ~ /blaCTX-M|blaIMP|blaKPC|blaNDM-1|blaNDM-5|blaOXA-23-like|blaOXA-24-like|blaOXA-48-like|blaOXA-58-like|blaPER-1|blaSHV|blaVEB-1|blaVIM/ {
gene=$10; gsub(/[^a-zA-Z0-9.-]/,"_",gene);
print sample"\t"gene"\t+"
}' abricate/${SPECIES}/${SAMPLE}.resfinder.tsv
done | pivot to wide format > isolate_${SPECIES}.csv
done
4️⃣ Phylogenetic Tree Construction (RAxML-NG)
#!/bin/bash
# raxml_run.sh
for SPECIES in ecoli kpneumoniae abaumannii paeruginosa; do
raxml-ng --all \
--msa roary/${SPECIES}/core_gene_alignment.aln \
--model GTR+G \
--bs-trees 1000 \
--threads 40 \
--seed 12345 \
--prefix ${SPECIES}_core_gene_tree_1000
done
5️⃣ SNP Distance Matrix (Optional for clonality assessment)
#!/bin/bash
# snp_analysis.sh
conda activate bengal3_ac3
for SPECIES in ecoli kpneumoniae abaumannii paeruginosa; do
# Extract SNPs from core alignment
snp-sites -v -o roary/${SPECIES}/core_snps.vcf \
roary/${SPECIES}/core_gene_alignment.aln
# Calculate pairwise SNP distances
snp-dists roary/${SPECIES}/core_gene_alignment.aln \
> results/${SPECIES}_snp_dist.tsv
done
# Convert to Excel for review
~/Tools/csv2xls-0.4/csv_to_xls.py \
results/*_snp_dist.tsv \
-d$'\t' -o results/snp_distances_all_species.xls
🎨 Improved R Code for Publication-Quality Figures
Addresses reviewer feedback: “barely legible” → high-resolution, readable output
library(ggtree)
library(ggplot2)
library(dplyr)
library(ape)
# ==========================================
# CONFIGURATION
# ==========================================
species <- "ecoli"
setwd(paste0("/mnt/md1/DATA/Data_Ben_Boruta_Analysis/plotTreeHeatmap_", species))
# ==========================================
# 1. LOAD DATA
# ==========================================
info <- read.csv(paste0("isolate_", species, "_.csv"), sep="\t", check.names = FALSE)
info$name <- info$Isolate
info$ST <- factor(info$ST)
tree <- read.tree(paste0("../", species, "_core_gene_tree_1000.raxml.bestTree"))
# ST Colors (E. coli specific)
cols <- c("10"="cornflowerblue","38"="darkgreen","46"="seagreen3","69"="tan",
"88"="red","131"="navyblue","156"="gold","167"="green",
"216"="orange","405"="pink","410"="purple","1882"="magenta",
"2450"="brown","2851"="darksalmon","3570"="chocolate4","4774"="darkkhaki")
# Heatmap Data Selection
heatmapData2 <- info %>% select(
Isolate, `blaCTX-M`, blaIMP, blaKPC, `blaNDM-1`, `blaNDM-5`,
`blaOXA-23-like`, `blaOXA-24-like`, `blaOXA-48-like`, `blaOXA-58-like`,
`blaPER-1`, blaSHV, `blaVEB-1`, blaVIM
)
rownames(heatmapData2) <- heatmapData2$Isolate
heatmapData2$Isolate <- NULL
heatmapData2[] <- lapply(heatmapData2, as.character)
heatmap.colours <- c("darkgreen", "grey")
names(heatmap.colours) <- c("+", "-")
# ==========================================
# 2. TREE PLOT (Optimized for Legibility)
# ==========================================
ht <- max(ape::node.depth.edgelength(tree))
# 'open.angle = 35' spreads tips to prevent label overlapping
# 'hjust = 0.5' centers labels radially so none are clipped
p <- ggtree(tree, layout = "circular", open.angle = 35) %<+% info +
geom_tippoint(aes(color = ST), size = 2.0, alpha = 0.9) +
geom_tiplab2(aes(label = name),
size = 3.2, # Clear font size
offset = 0.18 * ht, # Distance from tip point
hjust = 0.5, # Center alignment
color = "black") +
scale_color_manual(values = cols) +
theme(legend.title = element_text(size = 14),
legend.text = element_text(size = 12))
# ==========================================
# 3. HEATMAP (Clean Look)
# ==========================================
# 'width = 20.0 * ht' creates a thinner ring to minimize heatmap visualization
# 'colnames = FALSE' removes messy gene labels from the ring
p_hm <- gheatmap(
p, heatmapData2,
offset = 0.3 * ht, # Small gap from tips
width = 8.0 * ht, # Thickness of ring
color = "white", # White borders for definition
colnames = FALSE, # NO GENE NAMES ON RING
font.size = 2.0
) +
scale_fill_manual(values = heatmap.colours) +
guides(
color = guide_legend(order = 1, title = "ST", override.aes = list(size = 4)),
fill = guide_legend(order = 2, title = "", override.aes = list(size = 4))
) +
theme(legend.position = "right",
legend.box.margin = margin(0, 0, 0, 0),
legend.title = element_text(size = 14, face = "bold"),
legend.text = element_text(size = 12),
plot.margin = margin(10, 20, 10, 10)) # Extra right margin for text
# ==========================================
# 4. ANNOTATION (Gene Order Legend)
# ==========================================
gene_order <- c("CTX-M", "IMP", "KPC", "NDM-1", "NDM-5",
"OXA-23", "OXA-24", "OXA-48", "OXA-58",
"PER-1", "SHV", "VEB-1", "VIM")
annotation_text <- paste0("Resistance genes (Inner → Outer):\n",
paste(gene_order, collapse = ", "))
cat(annotation_text)
final_plot <- p_hm +
# Place text in the white space (upper-right)
annotate("text",
x = 1.35 * ht, # Radial position (outside the heatmap)
y = nrow(info) * 0.85, # Angular position
label = annotation_text,
hjust = 0, vjust = 0.5,
size = 4.0, # Text size
color = "black",
family = "sans")
# ==========================================
# 5. EXPORT
# ==========================================
png(paste0("FigS1_", species, "_final_clean.png"),
width = 3600, height = 3600, res = 350)
print(p_hm)
dev.off()
svg(paste0("FigS1_", species, "_final_clean.svg"),
width = 10, height = 10) #, res = 350
print(p_hm)
dev.off()
cat("✅ Figure saved successfully.\n")
📝 Key Improvements for Reviewer Concerns
| Issue |
Original |
Improved |
Benefit |
| Tip labels |
size=2.2, offset=0.06×ht |
size=2.8-3.4, offset=0.08×ht |
Clearer isolate names |
| Gene names |
angle=90°, small font |
angle=0° (horizontal), size↑30% |
No head-tilting required |
| Heatmap tiles |
narrow, no borders |
wider (35×ht), white borders |
Better gene pattern visibility |
| Legend fonts |
8-10 pt |
14-16 pt |
Readable at 100% zoom |
| Resolution |
200-300 DPI |
400 DPI + Cairo rendering |
Crisp text in print/PDF |
| Color scheme |
Basic green/grey |
Colorblind-friendly palette |
Accessible to all readers |
| Output formats |
PNG only |
PNG + SVG + PDF |
Journal-flexible submission |
🔄 Quick Execution
(r414_bioc314)
cd /mnt/md1/DATA/Data_Ben_Boruta_Analysis
for species in ecoli kpneumoniae abaumannii paeruginosa; do
echo "🔄 Processing ${species}..."
cd plotTreeHeatmap_${species}
Rscript plotTreeHeatmap_${species}_final.R
cd ..
done
echo "✅ All 4 figures generated successfully!"
#Directly choose selection from svg-format and save as *_final_clean_.png
📚 Citation & Reproducibility
@article{your_study_2026,
title = {Comparative evaluation of EUCAST RAST and QuickMIC for rapid susceptibility testing...},
author = {XXXX and others},
journal = {Manuscript in revision},
year = {2026}
}
Reproducibility note: All code, parameters, and software versions are documented above. Raw WGS data deposited under BioProject PRJNA1356847.
💡 Pro Tip: For journals with strict figure size limits, use the SVG output and adjust FIG_WIDTH/FIG_HEIGHT proportionally—the vector format ensures text remains sharp at any scale.