-
Preparing raw data for the batch 2 dataset
They are wildtype strains grown in different medium. Urine - human urine AUM - artificial urine medium MHB - Mueller-Hinton broth Urine(人类尿液):pH值、比重、温度、污染物、化学成分、微生物负荷。 AUM(人工尿液培养基):pH值、营养成分、无菌性、渗透压、温度、污染物。 MHB(Mueller-Hinton培养基):pH值、无菌性、营养成分、温度、渗透压、抗生素浓度。 阿奇霉素(Azithromycin,常缩写 AZI)是大环内酯类抗生素的一种。 * 作用机制:主要通过结合细菌核糖体 50S 亚基(23S rRNA),阻止蛋白质合成中的“肽链延伸”,从而抑制细菌生长(多为抑菌作用,某些情况下也可杀菌)。 * 常见适应证:上呼吸道/下呼吸道感染、支原体/衣原体感染、部分皮肤软组织感染等(具体要看地区指南和耐药情况)。 * 特点:半衰期较长、组织分布好,所以常见给药方案是“三日疗法/五日疗法”。但也因为广泛使用,耐药问题比较突出。 * 耐药机制(概念性):常见包括 1. 23S rRNA 甲基化(erm 基因)导致结合位点改变; 2. 外排泵增加(efflux); 3. 核糖体蛋白突变等。 * 注意事项(概念性):可能引起胃肠道不适;少数人有心电图 QT 间期延长风险;和某些药物相互作用需要注意(具体用药应遵医嘱)。 mkdir raw_data; cd raw_data # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-1/AUM-1_1.fq.gz AUM_r4_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-1/AUM-1_2.fq.gz AUM_r4_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-2/AUM-2_1.fq.gz AUM_r5_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-2/AUM-2_2.fq.gz AUM_r5_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-3/AUM-3_1.fq.gz AUM_r6_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/AUM-3/AUM-3_2.fq.gz AUM_r6_R2.fq.gz # # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-1/MHB-1_1.fq.gz MH_r4_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-1/MHB-1_2.fq.gz MH_r4_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-2/MHB-2_1.fq.gz MH_r5_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-2/MHB-2_2.fq.gz MH_r5_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-3/MHB-3_1.fq.gz MH_r6_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/MHB-3/MHB-3_2.fq.gz MH_r6_R2.fq.gz # # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-1/Urine-1_1.fq.gz Urine_r4_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-1/Urine-1_2.fq.gz Urine_r4_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-2/Urine-2_1.fq.gz Urine_r5_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-2/Urine-2_2.fq.gz Urine_r5_R2.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-3/Urine-3_1.fq.gz Urine_r6_R1.fq.gz # ln -s ../X101SC24105589-Z01-J001/01.RawData/Urine-3/Urine-3_2.fq.gz Urine_r6_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-1/AUM-1_1.fq.gz AUM_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-1/AUM-1_2.fq.gz AUM_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-2/AUM-2_1.fq.gz AUM_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-2/AUM-2_2.fq.gz AUM_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-3/AUM-3_1.fq.gz AUM_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-3/AUM-3_2.fq.gz AUM_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-1/AUM-AZI-1_1.fq.gz AUM-AZI_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-1/AUM-AZI-1_2.fq.gz AUM-AZI_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-2/AUM-AZI-2_1.fq.gz AUM-AZI_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-2/AUM-AZI-2_2.fq.gz AUM-AZI_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-3/AUM-AZI-3_1.fq.gz AUM-AZI_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/AUM-AZI-3/AUM-AZI-3_2.fq.gz AUM-AZI_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-1/MH-1_1.fq.gz MH_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-1/MH-1_2.fq.gz MH_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-2/MH-2_1.fq.gz MH_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-2/MH-2_2.fq.gz MH_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-3/MH-3_1.fq.gz MH_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-3/MH-3_2.fq.gz MH_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-1/MH-AZI-1_1.fq.gz MH-AZI_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-1/MH-AZI-1_2.fq.gz MH-AZI_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-2/MH-AZI-2_1.fq.gz MH-AZI_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-2/MH-AZI-2_2.fq.gz MH-AZI_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-3/MH-AZI-3_1.fq.gz MH-AZI_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/MH-AZI-3/MH-AZI-3_2.fq.gz MH-AZI_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-1/Urine-1_1.fq.gz Urine_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-1/Urine-1_2.fq.gz Urine_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-2/Urine-2_1.fq.gz Urine_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-2/Urine-2_2.fq.gz Urine_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-3/Urine-3_1.fq.gz Urine_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-3/Urine-3_2.fq.gz Urine_r3_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-1/Urine-AZI-1_1.fq.gz Urine-AZI_r1_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-1/Urine-AZI-1_2.fq.gz Urine-AZI_r1_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-2/Urine-AZI-2_1.fq.gz Urine-AZI_r2_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-2/Urine-AZI-2_2.fq.gz Urine-AZI_r2_R2.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-3/Urine-AZI-3_1.fq.gz Urine-AZI_r3_R1.fq.gz ln -s ../X101SC25062155-Z01-J002/01.RawData/Urine-AZI-3/Urine-AZI-3_2.fq.gz Urine-AZI_r3_R2.fq.gz -
Preparing the directory trimmed
mkdir trimmed trimmed_unpaired; for sample_id in AUM_r1 AUM_r2 AUM_r3 AUM_r4 AUM_r5 AUM_r6 Urine_r1 Urine_r2 Urine_r3 Urine_r4 Urine_r5 Urine_r6 MH_r1 MH_r2 MH_r3 MH_r4 MH_r5 MH_r6 AUM-AZI_r1 AUM-AZI_r2 AUM-AZI_r3 Urine-AZI_r1 Urine-AZI_r2 Urine-AZI_r3 MH-AZI_r1 MH-AZI_r2 MH-AZI_r3; do \ java -jar /home/jhuang/Tools/Trimmomatic-0.36/trimmomatic-0.36.jar PE -threads 100 raw_data/${sample_id}_R1.fq.gz raw_data/${sample_id}_R2.fq.gz trimmed/${sample_id}_R1.fq.gz trimmed_unpaired/${sample_id}_R1.fq.gz trimmed/${sample_id}_R2.fq.gz trimmed_unpaired/${sample_id}_R2.fq.gz ILLUMINACLIP:/home/jhuang/Tools/Trimmomatic-0.36/adapters/TruSeq3-PE-2.fa:2:30:10:8:TRUE LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 AVGQUAL:20; done 2> trimmomatic_pe.log; done -
Preparing samplesheet.csv
sample,fastq_1,fastq_2,strandedness Urine_r1,Urine_r1_R1.fq.gz,Urine_r1_R2.fq.gz,auto Urine_r2,Urine_r2_R1.fq.gz,Urine_r2_R2.fq.gz,auto Urine_r3,Urine_r3_R1.fq.gz,Urine_r3_R2.fq.gz,auto Urine_r4,Urine_r4_R1.fq.gz,Urine_r4_R2.fq.gz,auto Urine_r5,Urine_r5_R1.fq.gz,Urine_r5_R2.fq.gz,auto Urine_r6,Urine_r6_R1.fq.gz,Urine_r6_R2.fq.gz,auto AUM_r1,AUM_r1_R1.fq.gz,AUM_r1_R2.fq.gz,auto AUM_r2,AUM_r2_R1.fq.gz,AUM_r2_R2.fq.gz,auto AUM_r3,AUM_r3_R1.fq.gz,AUM_r3_R2.fq.gz,auto AUM_r4,AUM_r4_R1.fq.gz,AUM_r4_R2.fq.gz,auto AUM_r5,AUM_r5_R1.fq.gz,AUM_r5_R2.fq.gz,auto AUM_r6,AUM_r6_R1.fq.gz,AUM_r6_R2.fq.gz,auto MH_r1,MH_r1_R1.fq.gz,MH_r1_R2.fq.gz,auto MH_r2,MH_r2_R1.fq.gz,MH_r2_R2.fq.gz,auto MH_r3,MH_r3_R1.fq.gz,MH_r3_R2.fq.gz,auto MH_r4,MH_r4_R1.fq.gz,MH_r4_R2.fq.gz,auto MH_r5,MH_r5_R1.fq.gz,MH_r5_R2.fq.gz,auto MH_r6,MH_r6_R1.fq.gz,MH_r6_R2.fq.gz,auto Urine-AZI_r1,Urine-AZI_r1_R1.fq.gz,Urine-AZI_r1_R2.fq.gz,auto Urine-AZI_r2,Urine-AZI_r2_R1.fq.gz,Urine-AZI_r2_R2.fq.gz,auto Urine-AZI_r3,Urine-AZI_r3_R1.fq.gz,Urine-AZI_r3_R2.fq.gz,auto AUM-AZI_r1,AUM-AZI_r1_R1.fq.gz,AUM-AZI_r1_R2.fq.gz,auto AUM-AZI_r2,AUM-AZI_r2_R1.fq.gz,AUM-AZI_r2_R2.fq.gz,auto AUM-AZI_r3,AUM-AZI_r3_R1.fq.gz,AUM-AZI_r3_R2.fq.gz,auto MH-AZI_r1,MH-AZI_r1_R1.fq.gz,MH-AZI_r1_R2.fq.gz,auto MH-AZI_r2,MH-AZI_r2_R1.fq.gz,MH-AZI_r2_R2.fq.gz,auto MH-AZI_r3,MH-AZI_r3_R1.fq.gz,MH-AZI_r3_R2.fq.gz,auto -
Preparing CP059040.fasta, CP059040_gene.gff3 and CP059040.bed and nextflow run
# -- DEBUG_1 (CDS --> exon in CP059040.gff) -- #Checking the record (see below) in results/genome/CP059040.gtf #In ./results/genome/CP059040.gtf e.g. "CP059040.1 Genbank transcript 1 1398 . + . transcript_id "gene-H0N29_00005"; gene_id "gene-H0N29_00005"; gene_name "dnaA"; Name "dnaA"; gbkey "Gene"; gene "dnaA"; gene_biotype "protein_coding"; locus_tag "H0N29_00005";" #--featurecounts_feature_type 'transcript' returns only the tRNA results #Since the tRNA records have "transcript and exon". In gene records, we have "transcript and CDS". replace the CDS with exon grep -P "\texon\t" CP059040.gff | sort | wc -l #96 grep -P "cmsearch\texon\t" CP059040.gff | wc -l #=10 ignal recognition particle sRNA small typ, transfer-messenger RNA, 5S ribosomal RNA grep -P "Genbank\texon\t" CP059040.gff | wc -l #=12 16S and 23S ribosomal RNA grep -P "tRNAscan-SE\texon\t" CP059040.gff | wc -l #tRNA 74 wc -l star_salmon/AUM_r3/quant.genes.sf #--featurecounts_feature_type 'transcript' results in 96 records! grep -P "\tCDS\t" CP059040.gff | wc -l #3701 sed 's/\tCDS\t/\texon\t/g' CP059040.gff > CP059040_m.gff grep -P "\texon\t" CP059040_m.gff | sort | wc -l #3797 # -- DEBUG_2: combination of 'CP059040_m.gff' and 'exon' results in ERROR, using 'transcript' instead! --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024/CP059040_m.gff" --featurecounts_feature_type 'transcript' # ---- SUCCESSFUL with directly downloaded gff3 and fasta from NCBI using docker after replacing 'CDS' with 'exon' ---- (host_env) mv trimmed/*.fastq.gz . (host_env) nextflow run nf-core/rnaseq -r 3.14.0 -profile docker \ --input samplesheet.csv --outdir results --fasta "/home/jhuang/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/CP059040.fasta" --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/CP059040_m.gff" -resume --max_cpus 90 --max_memory 900.GB --max_time 2400.h --save_align_intermeds --save_unaligned --save_reference --aligner ‘star_salmon’ --gtf_group_features ‘gene_id’ --gtf_extra_attributes ‘gene_name’ --featurecounts_group_type ‘gene_biotype’ --featurecounts_feature_type ‘transcript’ # -- DEBUG_3: make sure the header of fasta is the same to the *_m.gff file -
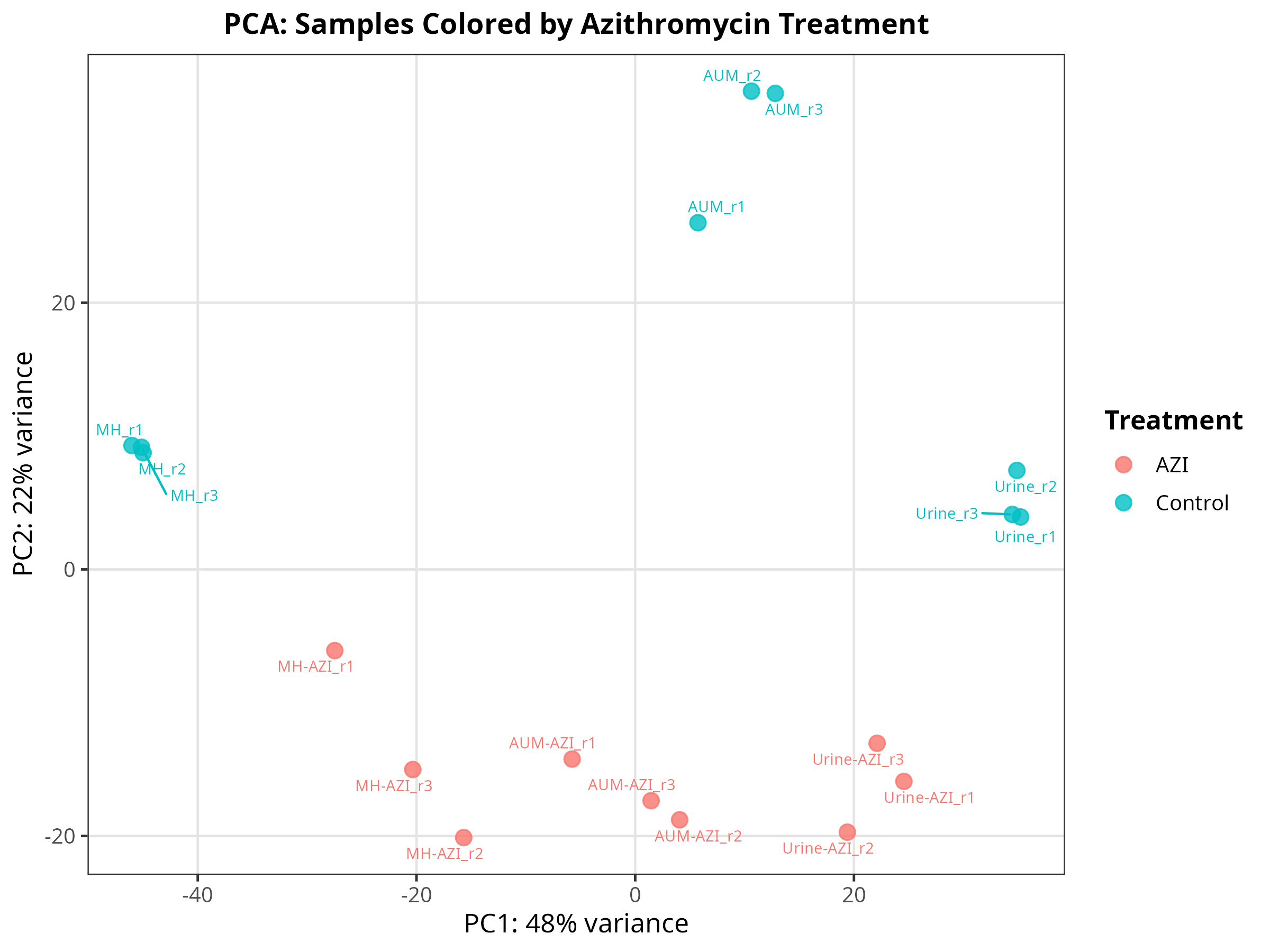
Import data and pca-plot
# ============================================================================== # ADAPTED PIPELINE: 6 Groups (Urine/AUM/MH ± AZI) -> Counts Export -> PCA # ============================================================================== # 1️⃣ LOAD LIBRARIES ------------------------------------------------------------ suppressPackageStartupMessages({ library(DESeq2) library(tximport) library(dplyr) library(ggplot2) library(ggrepel) library(edgeR) # For robust CPM calculation library(openxlsx) # For Excel export }) # 2️⃣ SET WORKING DIRECTORY & DEFINE SAMPLES ------------------------------------ setwd("/mnt/md1/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_on_ATCC19606/results/star_salmon") files <- c( "AUM-AZI_r1" = "./AUM-AZI_r1/quant.sf", "AUM-AZI_r2" = "./AUM-AZI_r2/quant.sf", "AUM-AZI_r3" = "./AUM-AZI_r3/quant.sf", "AUM_r1" = "./AUM_r1/quant.sf", "AUM_r2" = "./AUM_r2/quant.sf", "AUM_r3" = "./AUM_r3/quant.sf", "MH-AZI_r1" = "./MH-AZI_r1/quant.sf", "MH-AZI_r2" = "./MH-AZI_r2/quant.sf", "MH-AZI_r3" = "./MH-AZI_r3/quant.sf", "MH_r1" = "./MH_r1/quant.sf", "MH_r2" = "./MH_r2/quant.sf", "MH_r3" = "./MH_r3/quant.sf", "Urine-AZI_r1" = "./Urine-AZI_r1/quant.sf", "Urine-AZI_r2" = "./Urine-AZI_r2/quant.sf", "Urine-AZI_r3" = "./Urine-AZI_r3/quant.sf", "Urine_r1" = "./Urine_r1/quant.sf", "Urine_r2" = "./Urine_r2/quant.sf", "Urine_r3" = "./Urine_r3/quant.sf" ) # 3️⃣ AUTOMATED METADATA PARSING ----------------------------------------------- # Dynamically extracts Media, Treatment, and Combined Group from filenames samples <- names(files) colData <- data.frame( media = factor(gsub("-.*", "", samples)), treatment = factor(ifelse(grepl("AZI", samples), "AZI", "Control")), group = factor(paste(gsub("-.*", "", samples), ifelse(grepl("AZI", samples), "AZI", "Control"), sep = "_")), replicate = as.numeric(gsub(".*r", "", samples)), row.names = samples, stringsAsFactors = FALSE ) # 4️⃣ IMPORT & SUMMARIZE TO GENE LEVEL ----------------------------------------- tx2gene <- read.table("salmon_tx2gene.tsv", header = FALSE, stringsAsFactors = FALSE) colnames(tx2gene) <- c("transcript_id", "gene_id", "gene_name") tx2gene_geneonly <- tx2gene[, c("transcript_id", "gene_id")] # Direct gene-level import (faster & standard for DESeq2) txi <- tximport(files, type = "salmon", tx2gene = tx2gene_geneonly, txOut = FALSE) # Build DESeq2 object dds <- DESeqDataSetFromTximport(txi, colData = colData, design = ~ group) # Optional: Pre-filter low-count genes (improves VST & PCA stability) keep <- rowSums(counts(dds) >= 10) >= 3 dds <- dds[keep, ] # 5️⃣ EXPORT RAW COUNTS & CPM ------------------------------------------------- counts_data <- as.data.frame(counts(dds, normalized = FALSE)) counts_data$gene_id <- rownames(counts_data) # Merge gene names tx2gene_unique <- unique(tx2gene[, c("gene_id", "gene_name")]) counts_data <- merge(counts_data, tx2gene_unique, by = "gene_id", all.x = TRUE) count_cols <- setdiff(colnames(counts_data), c("gene_id", "gene_name")) counts_data <- counts_data[, c("gene_id", "gene_name", count_cols)] # Calculate CPM (edgeR handles library size normalization automatically) cpm_matrix <- edgeR::cpm(as.matrix(counts_data[, count_cols])) cpm_counts <- cbind(counts_data[, c("gene_id", "gene_name")], as.data.frame(cpm_matrix)) # Save tables write.csv(counts_data, "gene_raw_counts.csv", row.names = FALSE) write.xlsx(counts_data, "gene_raw_counts.xlsx", row.names = FALSE) write.xlsx(cpm_counts, "gene_cpm_counts.xlsx", row.names = FALSE) cat("✅ Count tables exported successfully.\n") # ============================================================================== # 6️⃣ PCA PLOTTING ------------------------------------------------------------- # ============================================================================== vsd <- vst(dds, blind = FALSE) pca_data <- plotPCA(vsd, intgroup = c("media", "treatment", "group"), returnData = TRUE) percent_var <- round(100 * attr(pca_data, "percentVar")) base_theme <- theme_bw(base_size = 12) + theme(plot.title = element_text(hjust = 0.5, face = "bold", size = 13), legend.position = "right", legend.title = element_text(face = "bold"), panel.grid.major = element_line(color = "grey90"), panel.grid.minor = element_blank()) # --- Plot 1: By Culture Media --- p1 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = media)) + geom_point(size = 3, alpha = 0.8) + geom_text_repel(aes(label = name), size = 2.5, max.overlaps = 20, show.legend = FALSE) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: Samples Colored by Media", color = "Media") + base_theme ggsave("01_PCA_by_Media.png", p1, width = 8, height = 6, dpi = 300) # --- Plot 2: By Treatment (AZI vs Control) --- p2 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = treatment)) + geom_point(size = 3, alpha = 0.8) + geom_text_repel(aes(label = name), size = 2.5, max.overlaps = 20, show.legend = FALSE) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: Samples Colored by Azithromycin Treatment", color = "Treatment") + base_theme ggsave("02_PCA_by_Treatment.png", p2, width = 8, height = 6, dpi = 300) # --- Plot 3: Combined Groups (Labeled) --- p3 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = group)) + geom_point(size = 3, alpha = 0.8) + geom_text_repel(aes(label = name), size = 2.2, max.overlaps = 30, box.padding = 0.3) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: Combined Media × Treatment Groups", color = "Group") + base_theme + theme(legend.position = "bottom") ggsave("03_PCA_CombinedGroups.png", p3, width = 9, height = 7, dpi = 300) # --- Plot 4: 95% Confidence Ellipses (by Media) --- p4 <- ggplot(pca_data, aes(x = PC1, y = PC2, color = media, fill = media)) + geom_point(size = 3, alpha = 0.7) + stat_ellipse(level = 0.95, alpha = 0.2, geom = "polygon", show.legend = FALSE) + labs(x = paste0("PC1: ", percent_var[1], "% variance"), y = paste0("PC2: ", percent_var[2], "% variance"), title = "PCA: 95% Confidence Ellipses by Media", color = "Media", fill = "Media") + base_theme ggsave("04_PCA_Ellipses.png", p4, width = 8, height = 6, dpi = 300) message("✅ All 4 PCA plots saved to working directory!") -
Perform DEG analysis
# In DESeq2, an NA in the pvalue column (and typically also in padj) is intentional and indicates that the gene was excluded from statistical testing. # Key Changes Made: # 1. NA Handling: Added pvalue = ifelse(is.na(pvalue), 1, pvalue) and padj = ifelse(is.na(padj), 1, padj) in the mutate block. This converts statistical NAs (usually from low counts or outliers) to 1, marking them as "Not Significant". # 2. Ordering: Genes with NA (now 1) will sort to the bottom of your Excel sheets and appear at y=0 on volcano plots, which is the correct visual representation for non-significant genes. # 3. Plot Safety: The padj_plot column still handles padj == 0 by converting it to 1e-305 to prevent -log10(0) = Inf errors in the volcano plot.
perform_DEG_analysis.R Rscript perform_DEG_analysis.R
- TODOs if requrested for KEGG and GO enrichments