-
Input data
#Data_Ute_smallRNA_3/ Data_Ute_smallRNA_4/ Data_Ute_smallRNA_7/ #Data_Ute_smallRNA_3_nextflow/ Data_Ute_smallRNA_6_size_selected/ Data_Ute_smallRNA_7_out/ See samples in Sequencing_overview.xlsx Batch,Date,Status 1,August 2021,⚪ Experimental (nf655, nf657) 2,February 2022,❌ Not found in my processed datasets (nf701, nf705) 3,August 2022,⚪ Experimental (nf774, nf780) 4,November 2022,⚪ Experimental (nf796, nf797) 5,June 2023,⚪ Experimental (nf876, nf887 (MKL-1 but scr_DMSO!)) 6,August 2023,⚪ Experimental (nf916, nf917, nf918 (MKL-1 but scr_DMSO!), nf919 (MKL-1 but scr_DMSO!)) #? Maybe we can include the parental cells, EVs (see 3 MKL-1_wt, 1 MKL_1_derived_EV_miRNA in the point 'BATCH_1, 3, 4') and scr_DMSO_EVs for MKL-1 (3 samples in BATCH_5 and BATCH_6) for exceRpt running! #BATCH_1: (plot-numpy1) jhuang@WS-2290C:/media/jhuang/Elements/Data_Ute/Data_Ute_smallRNA_3$ find . -name "*fastq.gz" (6 samples) ./210817_NB501882_0294_AHW5Y2BGXJ/nf656/MKL_1_derived_EV_RNA_S13_R1_001.fastq.gz ./210817_NB501882_0294_AHW5Y2BGXJ/nf658/WaGa_derived_EV_RNA_S14_R1_001.fastq.gz #ERROR_wrong_demulti ./210817_NB501882_0294_AHW5Y2BGXJ_neu/nf655/MKL_1_derived_EV_miRNA_S1_R1_001.fastq.gz #ERROR_wrong_demulti ./210817_NB501882_0294_AHW5Y2BGXJ_neu/nf657/WaGa_derived_EV_miRNA_S2_R1_001.fastq.gz #BATCH_3: ./220617_NB501882_0371_AH7572BGXM/nf774/0403_WaGa_wt_S20_R1_001.fastq.gz #* ./220617_NB501882_0371_AH7572BGXM/nf780/0505_MKL1_wt_S26_R1_001.fastq.gz # newDemulti of BATCH_1, 3, 4: * ./230623_newDemulti_smallRNAs/210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti/2021_nf_ute_smallRNA/nf655/MKL_1_derived_EV_miRNA_S1_R1_001.fastq.gz * * ./230623_newDemulti_smallRNAs/210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti/2021_nf_ute_smallRNA/nf657/WaGa_derived_EV_miRNA_S2_R1_001.fastq.gz * ./230623_newDemulti_smallRNAs/220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf774/0403_WaGa_wt_S1_R1_001.fastq.gz * ./230623_newDemulti_smallRNAs/220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf780/0505_MKL1_wt_S2_R1_001.fastq.gz * * ./230623_newDemulti_smallRNAs/221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf796/MKL-1_wt_1_S1_R1_001.fastq.gz * * ./230623_newDemulti_smallRNAs/221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf797/MKL-1_wt_2_S2_R1_001.fastq.gz * #BATCH_5: (plot-numpy1) jhuang@WS-2290C:/media/jhuang/Elements/Data_Ute/Data_Ute_smallRNA_4$ find . -name "*.fastq.gz" () #ERROR_wrong_adapter ./230602_NB501882_0428_AHKG53BGXT/demulti_wrong_adapter_trimming/nf876/1002_WaGa_sT_Dox_S1_R1_001.fastq.gz #ERROR_wrong_adapter ./230602_NB501882_0428_AHKG53BGXT/demulti_wrong_adapter_trimming/nf887/2312_MKL_1_scr_DMSO_S2_R1_001.fastq.gz ./230602_NB501882_0428_AHKG53BGXT/demulti_new/nf876/1002_WaGa_sT_Dox_S1_R1_001.fastq.gz * NOT_USED: MKL-1 but scr_DMSO: ./230602_NB501882_0428_AHKG53BGXT/demulti_new/nf887/2312_MKL_1_scr_DMSO_S2_R1_001.fastq.gz #BATCH_6: (plot-numpy1) jhuang@WS-2290C:/media/jhuang/Elements/Data_Ute/Data_Ute_smallRNA_6_size_selected$ find . -name "*.fastq.gz" ./230811_NB501882_0432_AHMW2CBGXT/nf916/1002_WaGa_wt_S1_R1_001.fastq.gz ./230811_NB501882_0432_AHMW2CBGXT/nf917/1002_WaGa_sT_Dox_S2_R1_001.fastq.gz * NOT_USED: MKL-1 but scr_DMSO: ./230811_NB501882_0432_AHMW2CBGXT/nf918/2312_MKL_1_scr_DMSO_S3_R1_001.fastq.gz * NOT_USED: MKL-1 but scr_DMSO: ./230811_NB501882_0432_AHMW2CBGXT/nf919/2403_MKL_1_scr_DMSO_S4_R1_001.fastq.gz # BATCH_100 # Data_Ute_smallRNA_7/$ find . -name "*fastq.gz" (10 samples) ./231016_NB501882_0435_AHG7HMBGXV/nf930/01_0505_WaGa_wt_EV_RNA_S1_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf931/02_0505_WaGa_sT_DMSO_EV_RNA_S2_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf932/03_0505_WaGa_sT_Dox_EV_RNA_S3_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf933/04_0505_WaGa_scr_DMSO_EV_RNA_S4_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf934/05_0505_WaGa_scr_Dox_EV_RNA_S5_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf935/06_1905_WaGa_wt_EV_RNA_S6_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf936/07_1905_WaGa_sT_DMSO_EV_RNA_S7_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf937/08_1905_WaGa_sT_Dox_EV_RNA_S8_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf938/09_1905_WaGa_scr_DMSO_EV_RNA_S9_R1_001.fastq.gz ./231016_NB501882_0435_AHG7HMBGXV/nf939/10_1905_WaGa_scr_Dox_EV_RNA_S10_R1_001.fastq.gz #I can’t find the nf940/11_control_MKL1_S11_R1_001.fastq.gz sample so I am not sure what kind of sample or condition it is. #NOT_USED_SINCE_IT_DOES_NOT_BELONG_TO_THE_PROJECT: ./231016_NB501882_0435_AHG7HMBGXV/nf940/11_control_MKL1_S11_R1_001.fastq.gz #NOT_USED_SINCE_IT_DOES_NOT_BELONG_TO_THE_PROJECT: ./231016_NB501882_0435_AHG7HMBGXV/nf941/12_control_WaGa_S12_R1_001.fastq.gz # Data_Ute_smallRNA_7$ find . -name "*.fastq.gz" (6 samples) ./250411_VH00358_135_AAGKGLHM5/nf961/WaGaWTcells_1_S1_R1_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf962/WaGaWTcells_2_S2_R1_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf971/2001_WaGa_sT_DMSO_S3_R1_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf972/2001_WaGa_sT_Dox_S4_R1_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf973/2001_WaGa_scr_DMSO_S5_R1_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf974/2001_WaGa_scr_Dox_S6_R1_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf961/WaGaWTcells_1_S1_R2_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf962/WaGaWTcells_2_S2_R2_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf971/2001_WaGa_sT_DMSO_S3_R2_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf972/2001_WaGa_sT_Dox_S4_R2_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf973/2001_WaGa_scr_DMSO_S5_R2_001.fastq.gz ./250411_VH00358_135_AAGKGLHM5/nf974/2001_WaGa_scr_Dox_S6_R2_001.fastq.gz #941,07 € 1051,79*0.85=894.0215 #FINAL DATASETS for running: newDemulti_of_BATCH_1_3_4 + BATCH_100 - Samples[nf941, nf940]!, Using directly with the orignal sample names nf*. MKL-1 wt cells (nf780*, nf796*, nf797*) MKL-1 wt_EV_RNA (nf655*) WaGa wt cells (nf774* (Considering to be deleted, due to possibly be an outlier, but in the current version, it is still included in the analysis), nf961, nf962) WaGa wt_EV_RNA (nf657* (The sample was excluded, since it is obviously a outlier, not clustered with the other 2 samples), nf930, nf935) WaGa_sT_DMSO_EV_RNA (nf931, nf936, nf971) WaGa_sT_Dox_EV_RNA (nf932, nf937, nf972) WaGa_scr_DMSO_EV_RNA (nf933, nf938, nf973) WaGa_scr_Dox_EV_RNA (nf934, nf939, nf974) -
Adapter trimming
#some common adapter sequences from different kits for reference: # - TruSeq Small RNA (Illumina): TGGAATTCTCGGGTGCCAAGG # - Small RNA Kits V1 (Illumina): TCGTATGCCGTCTTCTGCTTGT # - Small RNA Kits V1.5 (Illumina): ATCTCGTATGCCGTCTTCTGCTTG # - NEXTflex Small RNA Sequencing Kit v3 for Illumina Platforms (Bioo Scientific): TGGAATTCTCGGGTGCCAAGG # - LEXOGEN Small RNA-Seq Library Prep Kit (Illumina): TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC * mkdir Data_Ute_exceRpt_workspace/trimmed; cd Data_Ute_exceRpt_workspace/trimmed echo "------------------------------------ cutadapting nf774 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf774.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf774/0403_WaGa_wt_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf657 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf657.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti/2021_nf_ute_smallRNA/nf657/WaGa_derived_EV_miRNA_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf655 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf655.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti/2021_nf_ute_smallRNA/nf655/MKL_1_derived_EV_miRNA_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf780 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf780.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf780/0505_MKL1_wt_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf796 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf796.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf796/MKL-1_wt_1_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf797 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf797.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf797/MKL-1_wt_2_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf930 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf930.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf930/01_0505_WaGa_wt_EV_RNA_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf931 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf931.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf931/02_0505_WaGa_sT_DMSO_EV_RNA_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf932 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf932.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf932/03_0505_WaGa_sT_Dox_EV_RNA_S3_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf933 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf933.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf933/04_0505_WaGa_scr_DMSO_EV_RNA_S4_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf934 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf934.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf934/05_0505_WaGa_scr_Dox_EV_RNA_S5_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf935 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf935.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf935/06_1905_WaGa_wt_EV_RNA_S6_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf936 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf936.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf936/07_1905_WaGa_sT_DMSO_EV_RNA_S7_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf937 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf937.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf937/08_1905_WaGa_sT_Dox_EV_RNA_S8_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf938 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf938.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf938/09_1905_WaGa_scr_DMSO_EV_RNA_S9_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf939 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf939.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf939/10_1905_WaGa_scr_Dox_EV_RNA_S10_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf940 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf940.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf940/11_control_MKL1_S11_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf941 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf941.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf941/12_control_WaGa_S12_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf961 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf961.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf961/WaGaWTcells_1_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf962 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf962.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf962/WaGaWTcells_2_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf971 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf971.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf971/2001_WaGa_sT_DMSO_S3_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf972 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf972.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf972/2001_WaGa_sT_Dox_S4_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf973 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf973.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf973/2001_WaGa_scr_DMSO_S5_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf974 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf974.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf974/2001_WaGa_scr_Dox_S6_R1_001.fastq.gz >> LOG -
Install exceRpt (https://github.gersteinlab.org/exceRpt/)
docker pull rkitchen/excerpt mkdir MyexceRptDatabase cd /mnt/nvme0n1p1/MyexceRptDatabase wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg38_lowmem.tgz tar -xvf exceRptDB_v4_hg38_lowmem.tgz #http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg19_lowmem.tgz #http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg38_lowmem.tgz #http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_mm10_lowmem.tgz wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_EXOmiRNArRNA.tgz tar -xvf exceRptDB_v4_EXOmiRNArRNA.tgz wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_EXOGenomes.tgz tar -xvf exceRptDB_v4_EXOGenomes.tgz -
Run exceRpt
#[---- REAL_RUNNING_COMPLETE_DB ---->] #NOTE that if not renamed in the input files, then have to RENAME all files recursively by removing "_cutadapted.fastq" in all names in _CORE_RESULTS_v4.6.3.tgz (first unzip, removing, then zip, mv to ../results_g). cd trimmed for file in *.fastq.gz; do echo "mv \"$file\" \"${file/.fastq/}\"" done mkdir results for sample in nf780 nf796 nf797 nf655 nf774 nf961 nf962 nf657 nf930 nf935 nf931 nf936 nf971 nf932 nf937 nf972 nf933 nf938 nf973 nf934 nf939 nf974; do docker run -v ~/DATA/Data_Ute_exceRpt_workspace/trimmed:/exceRptInput \ -v ~/DATA/Data_Ute_exceRpt_workspace/results:/exceRptOutput \ -v /mnt/nvme0n1p1/MyexceRptDatabase:/exceRpt_DB \ -t rkitchen/excerpt \ INPUT_FILE_PATH=/exceRptInput/${sample}.gz MAIN_ORGANISM_GENOME_ID=hg38 N_THREADS=50 JAVA_RAM='200G' MAP_EXOGENOUS=on done #DEBUG the excerpt env docker inspect rkitchen/excerpt:latest # Without /bin/bash → May run and exit immediately #docker run -it rkitchen/excerpt # With /bin/bash → Stays open for interaction docker run -it --entrypoint /bin/bash rkitchen/excerpt -
Processing exceRpt output from multiple samples
cd ~/DATA/Data_Ute_exceRpt_workspace/exceRpt-master mamba activate r_env mamba install -c conda-forge -c bioconda \ bioconductor-marray \ bioconductor-rgraphviz \ r-plyr r-gplots r-reshape2 r-ggplot2 r-scales r-openxlsx r-rcurl r-xml \ -y mamba install -c conda-forge -c bioconda \ r-plyr r-gplots r-reshape2 r-ggplot2 r-scales r-openxlsx \ bioconductor-marray bioconductor-rgraphviz \ -y #EXCLUDE some isolates since they have total different pattern --> outliner, e.g. nf657 from WaGa wt EV: sudo mv nf657* ../results_EXCLUDED/ mkdir summaries summariesWithSampleGroupInfo (r_env) jhuang@WS-2290C:~/DATA/Data_Ute_exceRpt_workspace/exceRpt-master$ R #WARNING: need to reload the R-script after each change of the script. source("mergePipelineRuns_functions.R") processSamplesInDir("../results/", "../summaries") #mkdir summariesWithSampleGroupInfo; cp summaries/*.RData summariesWithSampleGroupInfo; rm summariesWithSampleGroupInfo/exceRpt_sampleGroupDefinitions.txt; source("mergePipelineRuns_functions_addSampleGroupInfo.R") processSamplesInDir("../results/", "../summariesWithSampleGroupInfo") #!!!!! IMPORTANT: REPORT summariesWithSampleGroupInfo/exceRpt_DiagnosticPlots.pdf !!!!! #POSSIBLE_TODO (MAYBE BETTER NOT DO THIS, SINCE I HAVE TO GENERATE PCA- and MANHATTAN PLOTS!!): Wir need to delete nf774 from WaGa_wt_cells, since it is very far to another two samples! #~/Tools/csv2xls-0.4/csv_to_xls.py exceRpt_miRNA_ReadsPerMillion.txt exceRpt_tRNA_ReadsPerMillion.txt exceRpt_piRNA_ReadsPerMillion.txt -d$'\t' -o exceRpt_results_detailed.xls -
Downstream analyis using R for miRNAs (4 MKL-1 + 17 WaGa samples)
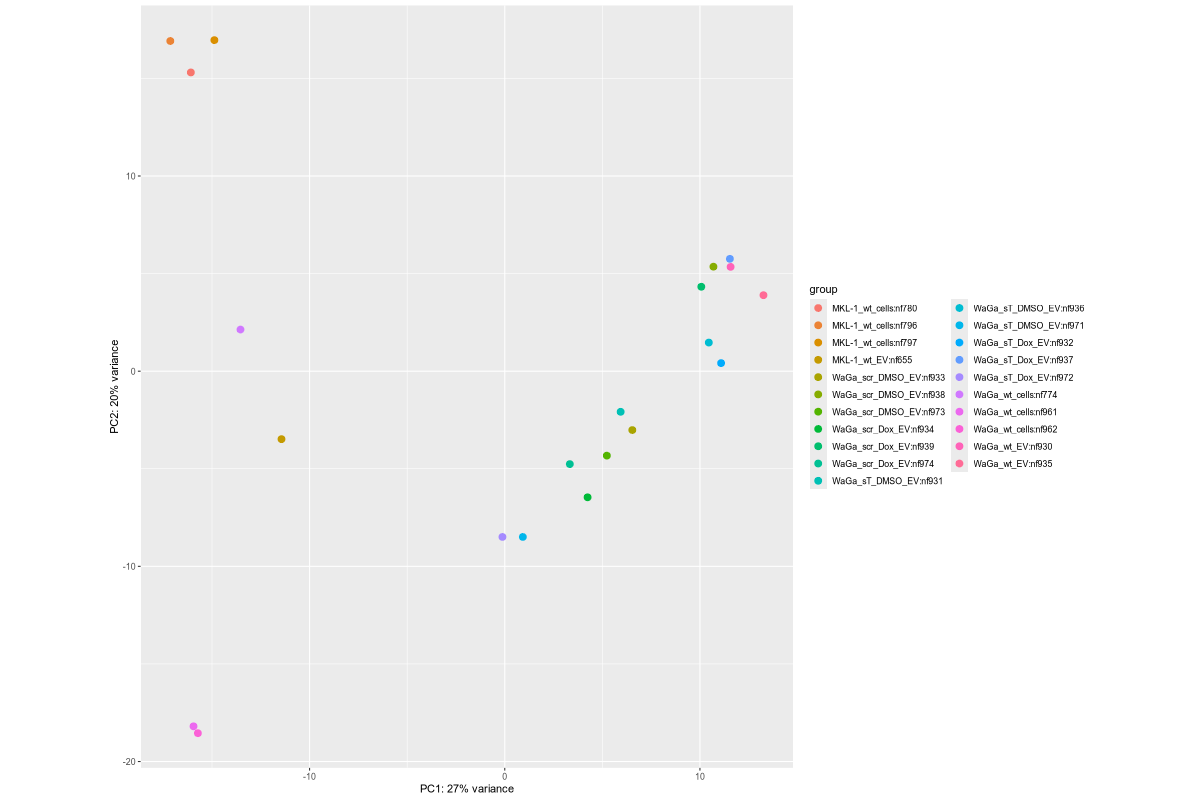
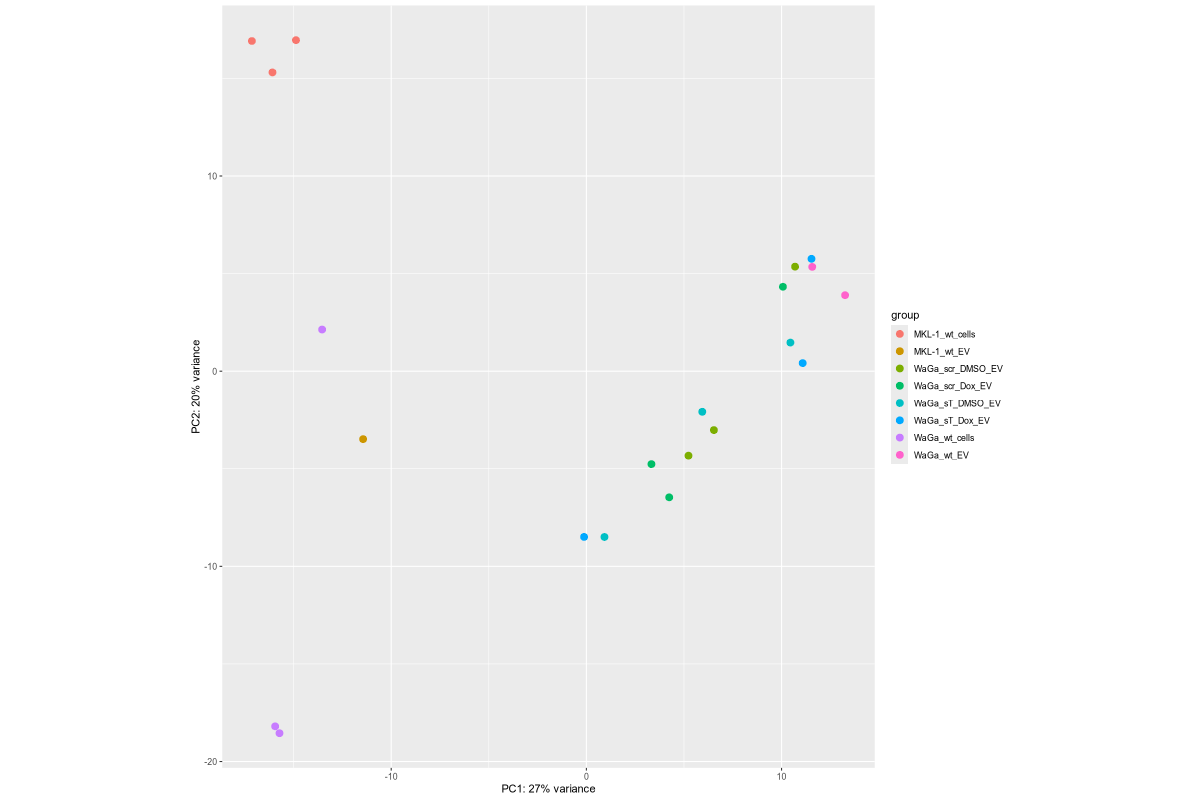
#Input file #exceRpt_miRNA_ReadCounts.txt #exceRpt_piRNA_ReadCounts.txt ## MKL-1 wild-type controls #MKL-1_wt_cells (nf780, nf796, nf797) #MKL-1_wt_EV (nf655) # ## WaGa experimental groups (scr = scramble control; sT = target knockdown) #WaGa_scr_DMSO_EV (nf933, nf938, nf973) #WaGa_scr_Dox_EV (nf934, nf939, nf974) #WaGa_sT_DMSO_EV (nf931, nf936, nf971) #WaGa_sT_Dox_EV (nf932, nf937, nf972) # ## WaGa wild-type controls #WaGa_wt_cells (nf774, nf961, nf962) #WaGa_wt_EV (nf657 (deleted), nf930, nf935) cd ~/DATA/Data_Ute_exceRpt_workspace/summaries mamba activate r_env R #> .libPaths() #[1] "/home/jhuang/mambaforge/envs/r_env/lib/R/library" #BiocManager::install("AnnotationDbi") #BiocManager::install("clusterProfiler") #BiocManager::install(c("ReactomePA","org.Hs.eg.db")) #BiocManager::install("limma") #BiocManager::install("sva") #install.packages("writexl") #install.packages("openxlsx") library("AnnotationDbi") library("clusterProfiler") library("ReactomePA") library("org.Hs.eg.db") library(DESeq2) library(gplots) library(limma) library(sva) #library(writexl) #d.raw_with_rownames <- cbind(RowNames = rownames(d.raw), d.raw); write_xlsx(d.raw, path = "d_raw.xlsx"); library(openxlsx) setwd("../summaries/") d.raw<- read.delim2("exceRpt_miRNA_ReadCounts.txt",sep="\t", header=TRUE, row.names=1) # Desired column order desired_order <- c( "nf780", "nf796", "nf797", "nf655", "nf933", "nf938", "nf973", "nf934", "nf939", "nf974", "nf931", "nf936", "nf971", "nf932", "nf937", "nf972", "nf774", "nf961", "nf962", "nf930", "nf935" #delete "nf657", ) # Reorder columns d.raw <- d.raw[, desired_order] setdiff(desired_order, colnames(d.raw)) # Shows missing or misnamed columns #sapply(d.raw, is.numeric) d.raw[] <- lapply(d.raw, as.numeric) #d.raw[] <- lapply(d.raw, function(x) as.numeric(as.character(x))) d.raw <- round(d.raw) write.csv(d.raw, file ="d_raw.csv") write.xlsx(d.raw, file = "d_raw.xlsx", rowNames = TRUE) # ------ Code sent to Ute ------ #d.raw <- read.delim2("d_raw.csv",sep=",", header=TRUE, row.names=1) Cell_or_EV = as.factor(c("Cell","Cell","Cell", "EV", "EV","EV","EV", "EV","EV","EV", "EV","EV","EV", "EV","EV","EV", "Cell","Cell","Cell", "EV","EV")) #delete "EV", replicates = as.factor(c("MKL-1_wt_cells","MKL-1_wt_cells","MKL-1_wt_cells", "MKL-1_wt_EV", "WaGa_scr_DMSO_EV","WaGa_scr_DMSO_EV","WaGa_scr_DMSO_EV", "WaGa_scr_Dox_EV","WaGa_scr_Dox_EV","WaGa_scr_Dox_EV", "WaGa_sT_DMSO_EV","WaGa_sT_DMSO_EV","WaGa_sT_DMSO_EV", "WaGa_sT_Dox_EV","WaGa_sT_Dox_EV","WaGa_sT_Dox_EV", "WaGa_wt_cells", "WaGa_wt_cells","WaGa_wt_cells", "WaGa_wt_EV", "WaGa_wt_EV")) #delete "WaGa_wt_EV", ids = as.factor(c("nf780", "nf796", "nf797", "nf655", "nf933", "nf938", "nf973", "nf934", "nf939", "nf974", "nf931", "nf936", "nf971", "nf932", "nf937", "nf972", "nf774", "nf961", "nf962", "nf930", "nf935")) #delete "nf657", cData = data.frame(row.names=colnames(d.raw), replicates=replicates, ids=ids, Cell_or_EV=Cell_or_EV) dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) # Filter low-count miRNAs dds <- dds[ rowSums(counts(dds)) > 10, ] #1322-->903 rld <- rlogTransformation(dds) # -- before pca -- png("pca.png", 1200, 800) plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) #plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() png("pca2.png", 1200, 800) #plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() # Batch Effect Removal Methods (Non-batch effect removal applied!) #### STEP2: DEGs #### #- Heatmap untreated/wt vs parental; 1x for WaGa cell line #- Volcano plot untreated/wt vs parental; 1x for WaGa cell line #- Manhattan plot miRNAs; 1x for WaGa cell line #- Distribution of different small RNA species untreated/wt and parental; 1x for WaGa cell line #- Motif analysis: identify RNA-binding proteins that may regulate small RNA loading; 1x for WaGa cell line #convert bam to bigwig using deepTools by feeding inverse of DESeq’s size Factor sizeFactors(dds) #NULL dds <- estimateSizeFactors(dds) sizeFactors(dds) normalized_counts <- counts(dds, normalized=TRUE) write.table(normalized_counts, file="normalized_counts.txt", sep="\t", quote=F, col.names=NA) write.xlsx(normalized_counts, file = "normalized_counts.xlsx", rowNames = TRUE) #---- untreated, scr_control, DMSO_control, scr_DMSO_control, sT_knockdown to parental_cells ---- dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) dds$replicates <- relevel(dds$replicates, "untreated") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("DMSO_control_vs_untreated", "scr_control_vs_untreated", "scr_DMSO_control_vs_untreated", "sT_knockdown_vs_untreated") dds$replicates <- relevel(dds$replicates, "DMSO_control") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_knockdown_vs_DMSO_control") dds$replicates <- relevel(dds$replicates, "scr_control") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_knockdown_vs_scr_control") dds$replicates <- relevel(dds$replicates, "scr_DMSO_control") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_knockdown_vs_scr_DMSO_control") dds$replicates <- relevel(dds$replicates, "WaGa_wt_cells") dds = DESeq(dds, betaPrior=FALSE) #default betaPrior is FALSE resultsNames(dds) clist <- c("WaGa_wt_EV_vs_WaGa_wt_cells") dds$replicates <- relevel(dds$replicates, "MKL-1_wt_cells") dds = DESeq(dds, betaPrior=FALSE) #default betaPrior is FALSE resultsNames(dds) clist <- c("MKL.1_wt_EV_vs_MKL.1_wt_cells") #NOTE that the results sent to Ute is |padj|<=0.1. for (i in clist) { contrast = paste("replicates", i, sep="_") res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] #https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#why-are-some-p-values-set-to-na res$padj <- ifelse(is.na(res$padj), 1, res$padj) res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(i, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(i, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(i, "down.txt", sep="-")) } ~/Tools/csv2xls-0.4/csv_to_xls.py \ untreated_vs_parental_cells-all.txt \ untreated_vs_parental_cells-up.txt \ untreated_vs_parental_cells-down.txt \ -d$',' -o untreated_vs_parental_cells.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ DMSO_control_vs_untreated-all.txt \ DMSO_control_vs_untreated-up.txt \ DMSO_control_vs_untreated-down.txt \ -d$',' -o DMSO_control_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ scr_control_vs_untreated-all.txt \ scr_control_vs_untreated-up.txt \ scr_control_vs_untreated-down.txt \ -d$',' -o scr_control_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ scr_DMSO_control_vs_untreated-all.txt \ scr_DMSO_control_vs_untreated-up.txt \ scr_DMSO_control_vs_untreated-down.txt \ -d$',' -o scr_DMSO_control_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_knockdown_vs_untreated-all.txt \ sT_knockdown_vs_untreated-up.txt \ sT_knockdown_vs_untreated-down.txt \ -d$',' -o sT_knockdown_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_knockdown_vs_DMSO_control-all.txt \ sT_knockdown_vs_DMSO_control-up.txt \ sT_knockdown_vs_DMSO_control-down.txt \ -d$',' -o sT_knockdown_vs_DMSO_control.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_knockdown_vs_scr_control-all.txt \ sT_knockdown_vs_scr_control-up.txt \ sT_knockdown_vs_scr_control-down.txt \ -d$',' -o sT_knockdown_vs_scr_control.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_knockdown_vs_scr_DMSO_control-all.txt \ sT_knockdown_vs_scr_DMSO_control-up.txt \ sT_knockdown_vs_scr_DMSO_control-down.txt \ -d$',' -o sT_knockdown_vs_scr_DMSO_control.xls; # ------------------- volcano_plot ------------------- library(ggplot2) library(ggrepel) geness_res <- read.csv(file = paste("untreated_vs_parental_cells", "all.txt", sep="-"), row.names=1) external_gene_name <- rownames(geness_res) geness_res <- cbind(geness_res, external_gene_name) #top_g are from ids top_g <- c("hsa-let-7b-5p","hsa-let-7g-5p","hsa-let-7i-5p","hsa-miR-103a-3p","hsa-miR-107","hsa-miR-1224-5p","hsa-miR-122-5p","hsa-miR-1226-5p","hsa-miR-1246","hsa-miR-127-3p","hsa-miR-1290","hsa-miR-130a-3p","hsa-miR-139-3p","hsa-miR-141-3p","hsa-miR-143-3p","hsa-miR-148b-3p","hsa-miR-155-5p","hsa-miR-15a-5p","hsa-miR-17-5p","hsa-miR-184","hsa-miR-18a-3p","hsa-miR-18a-5p","hsa-miR-190a-5p","hsa-miR-191-5p","hsa-miR-193b-5p","hsa-miR-197-5p","hsa-miR-200a-3p","hsa-miR-200b-5p","hsa-miR-206","hsa-miR-20a-5p","hsa-miR-210-3p","hsa-miR-2110","hsa-miR-21-5p","hsa-miR-218-5p","hsa-miR-219a-1-3p","hsa-miR-221-3p","hsa-miR-23b-3p","hsa-miR-27a-3p","hsa-miR-27b-3p","hsa-miR-27b-5p","hsa-miR-28-3p","hsa-miR-30a-5p","hsa-miR-30c-5p","hsa-miR-30e-5p","hsa-miR-3127-5p","hsa-miR-3131","hsa-miR-3180|hsa-miR-3180-3p","hsa-miR-320a","hsa-miR-320b","hsa-miR-320c","hsa-miR-320d","hsa-miR-330-3p","hsa-miR-335-3p","hsa-miR-33b-5p","hsa-miR-340-5p","hsa-miR-342-5p","hsa-miR-3605-5p","hsa-miR-361-3p","hsa-miR-365a-5p","hsa-miR-374b-5p","hsa-miR-378i","hsa-miR-379-5p","hsa-miR-3940-5p","hsa-miR-409-3p","hsa-miR-411-5p","hsa-miR-423-3p","hsa-miR-423-5p","hsa-miR-4286","hsa-miR-429","hsa-miR-432-5p","hsa-miR-4326","hsa-miR-451a","hsa-miR-4520-3p","hsa-miR-454-3p","hsa-miR-4646-5p","hsa-miR-4667-5p","hsa-miR-4748","hsa-miR-483-5p","hsa-miR-486-5p","hsa-miR-5010-5p","hsa-miR-504-3p","hsa-miR-5187-5p","hsa-miR-590-3p","hsa-miR-6128","hsa-miR-625-5p","hsa-miR-6726-5p","hsa-miR-6730-5p","hsa-miR-676-3p","hsa-miR-6767-5p","hsa-miR-6777-5p","hsa-miR-6780a-5p","hsa-miR-6794-5p","hsa-miR-6817-3p","hsa-miR-708-5p","hsa-miR-7-5p","hsa-miR-766-5p","hsa-miR-7854-3p","hsa-miR-873-3p","hsa-miR-885-3p","hsa-miR-92b-5p","hsa-miR-93-5p","hsa-miR-937-3p","hsa-miR-9-5p","hsa-miR-98-5p") subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)) geness_res$Color <- "NS or log2FC < 2.0" geness_res$Color[geness_res$pvalue < 0.05] <- "P < 0.05" geness_res$Color[geness_res$padj < 0.05] <- "P-adj < 0.05" geness_res$Color[abs(geness_res$log2FoldChange) < 2.0] <- "NS or log2FC < 2.0" write.csv(geness_res, "untreated_vs_parental_cells_with_Category.csv") geness_res$invert_P <- (-log10(geness_res$pvalue)) * sign(geness_res$log2FoldChange) geness_res <- geness_res[, -1*ncol(geness_res)] png("volcano_plot_untreated_vs_parental_cells.png",width=1200, height=1400) #svg("untreated_vs_parental_cells.svg",width=12, height=14) ggplot(geness_res, aes(x = log2FoldChange, y = -log10(pvalue), color = Color, label = external_gene_name)) + geom_vline(xintercept = c(2.0, -2.0), lty = "dashed") + geom_hline(yintercept = -log10(0.05), lty = "dashed") + geom_point() + labs(x = "log2(FC)", y = "Significance, -log10(P)", color = "Significance") + scale_color_manual(values = c("P < 0.05"="orange","P-adj < 0.05"="red","NS or log2FC < 2.0"="darkgray"),guide = guide_legend(override.aes = list(size = 4))) + scale_y_continuous(expand = expansion(mult = c(0,0.05))) + geom_text_repel(data = subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)), size = 4, point.padding = 0.15, color = "black", min.segment.length = .1, box.padding = .2, lwd = 2) + theme_bw(base_size = 16) + theme(legend.position = "bottom") dev.off() # ------------------ differentially_expressed_miRNAs_heatmap ----------------- # prepare all_genes rld <- rlogTransformation(dds) mat <- assay(rld) mm <- model.matrix(~replicates, colData(rld)) mat <- limma::removeBatchEffect(mat, batch=rld$batch, design=mm) assay(rld) <- mat RNASeq.NoCellLine <- assay(rld) # reorder the columns #colnames(RNASeq.NoCellLine) = c("0505 WaGa sT DMSO","1905 WaGa sT DMSO","0505 WaGa sT Dox","1905 WaGa sT Dox","0505 WaGa scr DMSO","1905 WaGa scr DMSO","0505 WaGa scr Dox","1905 WaGa scr Dox","0505 WaGa wt","1905 WaGa wt","control MKL1","control WaGa") #col.order <-c("control MKL1", "control WaGa","0505 WaGa wt","1905 WaGa wt","0505 WaGa sT DMSO","1905 WaGa sT DMSO","0505 WaGa sT Dox","1905 WaGa sT Dox","0505 WaGa scr DMSO","1905 WaGa scr DMSO","0505 WaGa scr Dox","1905 WaGa scr Dox") #RNASeq.NoCellLine <- RNASeq.NoCellLine[,col.order] #Option4: manully defining #for i in untreated_vs_parental_cells sT_knockdown_vs_untreated DMSO_control_vs_untreated scr_control_vs_untreated scr_DMSO_control_vs_untreated sT_knockdown_vs_DMSO_control sT_knockdown_vs_scr_control sT_knockdown_vs_scr_DMSO_control; do # echo "cut -d',' -f1-1 ${i}-up.txt > ${i}-up.id"; # echo "cut -d',' -f1-1 ${i}-down.txt > ${i}-down.id"; #done #cat *.id | sort -u > ids ##add Gene_Id in the first line, delete the "" GOI <- read.csv("ids")$Gene_Id datamat = RNASeq.NoCellLine[GOI, ] # clustering the genes and draw heatmap #datamat <- datamat[,-1] #delete the sample "control MKL1" #datamat <- datamat[, 1:5] #parental_cells_1 parental_cells_2 parental_cells_3 untreated_1 untreated_2 scr_control_1 scr_control_2 scr_control_3 DMSO_control_1 DMSO_control_2 DMSO_control_3 scr_DMSO_control_1 scr_DMSO_control_2 scr_DMSO_control_3 sT_knockdown_1 sT_knockdown_2 sT_knockdown_3 --> #parental cells 1 parental cells 2 parental cells 3 untreated 1 untreated 2 scr control 1 scr control 2 scr control 3 DMSO control 1 DMSO control 2 DMSO control 3 scr DMSO control 1 scr DMSO control 2 scr DMSO control 3 sT knockdown 1 sT knockdown 2 sT knockdown 3 colnames(datamat)[1] <- "parental cells 1" colnames(datamat)[2] <- "parental cells 2" colnames(datamat)[3] <- "parental cells 3" colnames(datamat)[4] <- "untreated 1" colnames(datamat)[5] <- "untreated 2" colnames(datamat)[6] <- "scr control 1" colnames(datamat)[7] <- "scr control 2" colnames(datamat)[8] <- "scr control 3" colnames(datamat)[9] <- "DMSO control 1" colnames(datamat)[10] <- "DMSO control 2" colnames(datamat)[11] <- "DMSO control 3" colnames(datamat)[12] <- "scr DMSO control 1" colnames(datamat)[13] <- "scr DMSO control 2" colnames(datamat)[14] <- "scr DMSO control 3" colnames(datamat)[15] <- "sT knockdown 1" colnames(datamat)[16] <- "sT knockdown 2" colnames(datamat)[17] <- "sT knockdown 3" write.csv(datamat, file ="gene_expression_keeping_replicates.txt") write.xlsx(datamat, file = "gene_expression_keeping_replicates.xlsx", rowNames = TRUE) #"ward.D"’, ‘"ward.D2"’,‘"single"’, ‘"complete"’, ‘"average"’ (= UPGMA), ‘"mcquitty"’(= WPGMA), ‘"median"’ (= WPGMC) or ‘"centroid"’ (= UPGMC) hr <- hclust(as.dist(1-cor(t(datamat), method="pearson")), method="complete") hc <- hclust(as.dist(1-cor(datamat, method="spearman")), method="complete") mycl = cutree(hr, h=max(hr$height)/1.1) mycol = c("YELLOW", "BLUE", "ORANGE", "CYAN", "GREEN", "MAGENTA", "GREY", "LIGHTCYAN", "RED", "PINK", "DARKORANGE", "MAROON", "LIGHTGREEN", "DARKBLUE", "DARKRED", "LIGHTBLUE", "DARKCYAN", "DARKGREEN", "DARKMAGENTA"); mycol = mycol[as.vector(mycl)] rownames(datamat) <- sub("\\|.*", "", rownames(datamat)) png("DEGs_heatmap_keeping_replicates.png", width=1000, height=1400) #svg("DEGs_heatmap_keeping_replicates.svg", width=6, height=8) heatmap.2(as.matrix(datamat), Rowv=as.dendrogram(hr), Colv=NA, dendrogram='row', labRow=row.names(datamat), scale='row', trace='none', col=bluered(75), RowSideColors=mycol, srtCol=30, lhei=c(1,8), cexRow=1.4, # Increase row label font size cexCol=1.7, # Increase column label font size margin=c(8, 12) ) dev.off() # ---------------------------------------- # ----------- manhattan_plot ------------- Rscript manhattan_plot_Carmen_custom_labels.R #exceRpt_miRNA_ReadCounts.txt
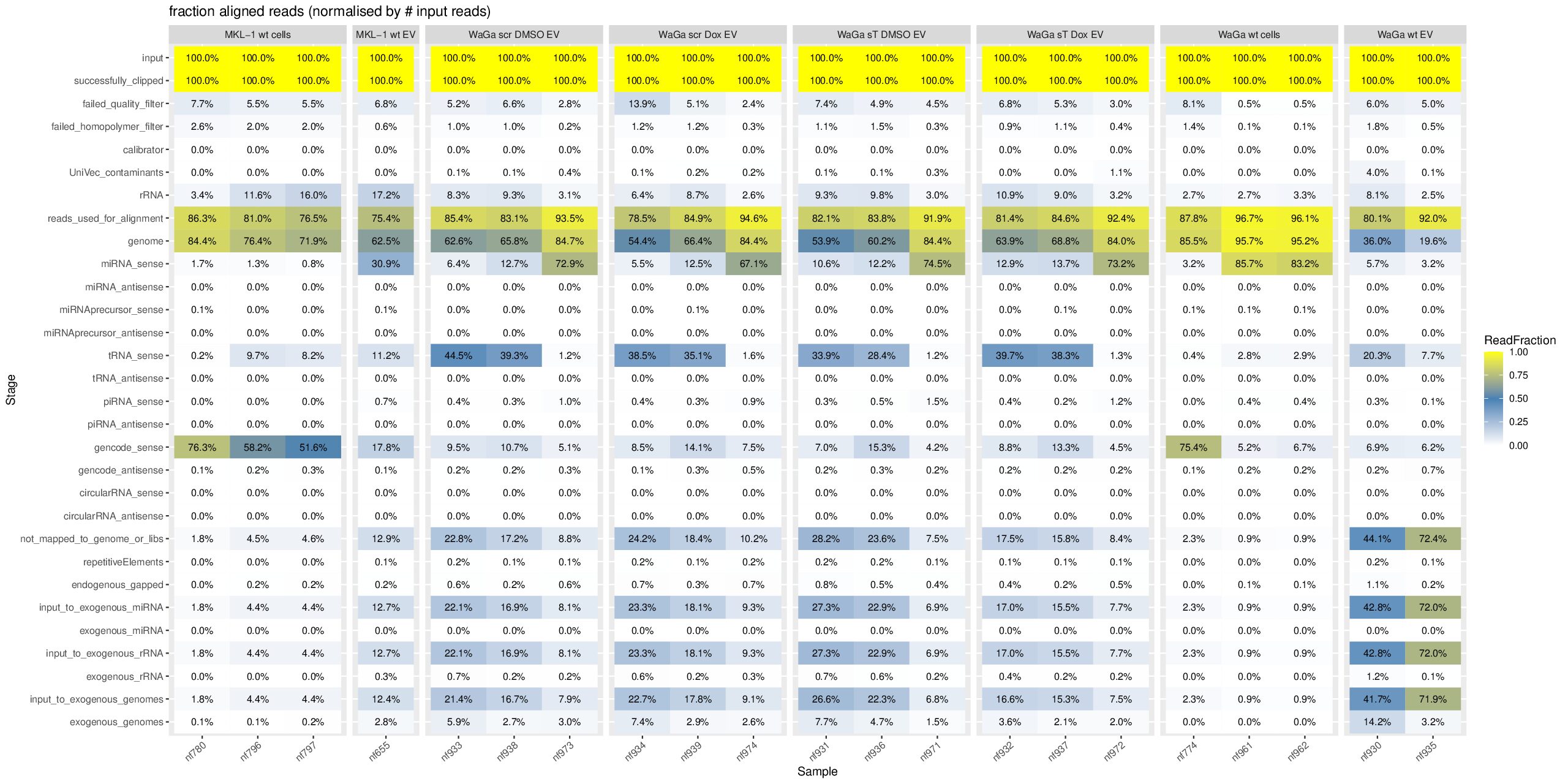
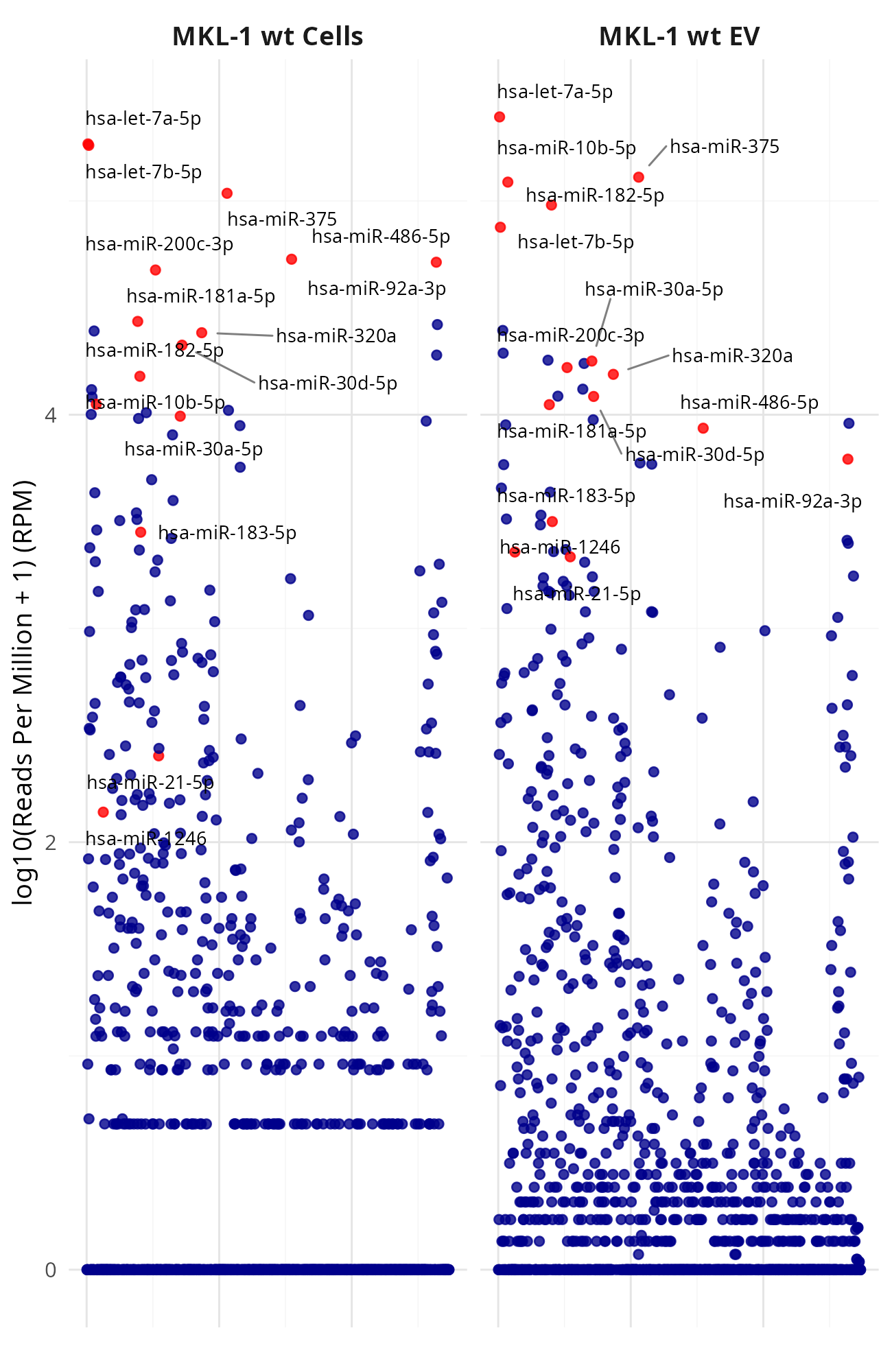
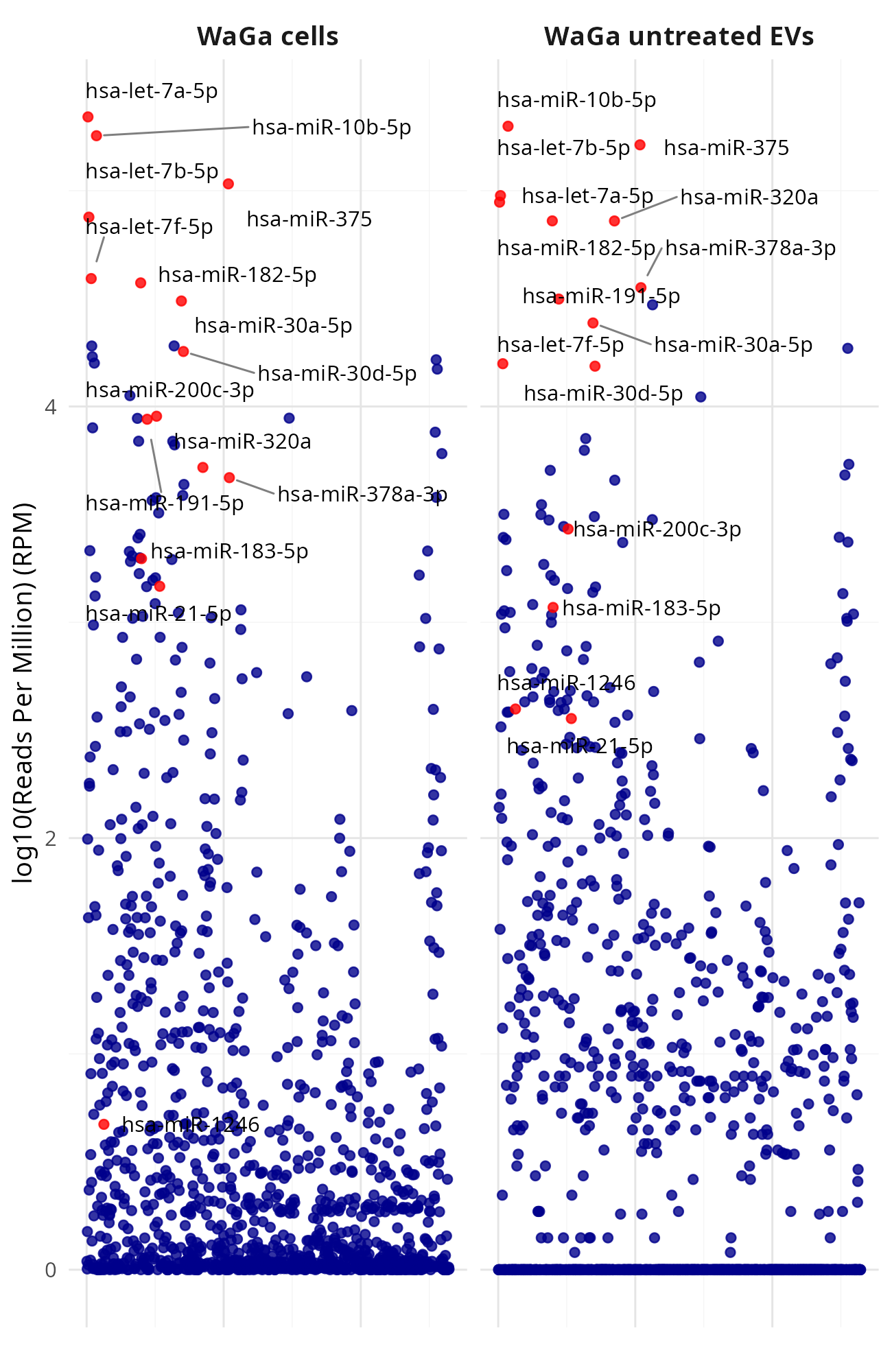
Generating manhattan plots for [MKL-1|WaGa] wt cells vs [MKL-1|WaGa] wt EV (Data_Ute_exceRpt_workspace)
Leave a reply