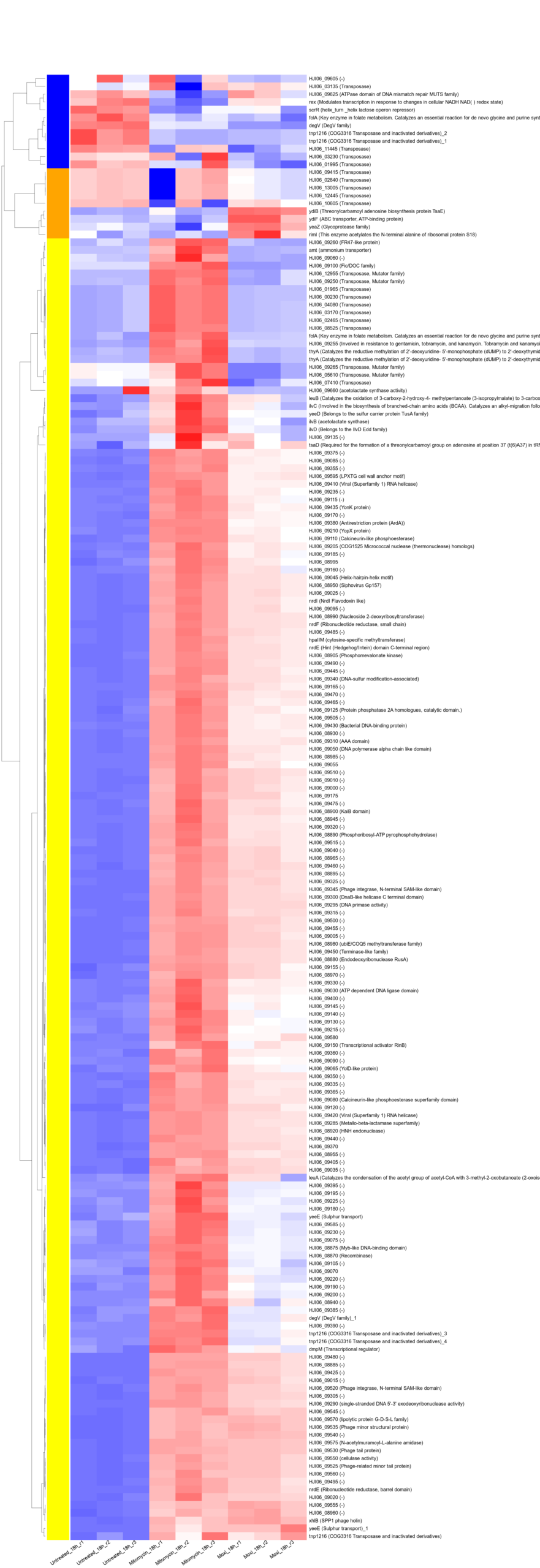
This post documents a complete, batch-ready pipeline to generate subset heatmaps (phage / stress-response / biofilm-associated) from bacterial RNA-seq data quantified with Salmon, using DE tables without any p-value cutoff.
You will end with:
-
Three gene sets (A/B/C):
- A (phage/prophage genes): extracted from MT880872.1.gb, mapped to CP052959 via BLASTN, converted to CP052959
GeneID_plain - B (stress genes): keyword-based selection from CP052959 GenBank annotations
- C (biofilm genes): keyword-based selection from CP052959 GenBank annotations
- A (phage/prophage genes): extracted from MT880872.1.gb, mapped to CP052959 via BLASTN, converted to CP052959
-
For each
*-all_annotated.csvinresults/star_salmon/degenes/:- Subset GOI lists for A/B/C (no cutoff; include all rows belonging to the geneset)
- Per-comparison
*_matched.tsvtables for sanity checks
- Merged 3-condition heatmaps (Untreated + Mitomycin + Moxi) per timepoint (4h/8h/18h) and subset (A/B/C), giving 9 final figures
- An Excel file per heatmap containing
GeneID,GeneName,Description, and the plotted expression matrix
Everything is written so you can run a single shell script for genesets + intersections, then one R script for heatmaps.
0) Environments
We use two conda environments:
plot-numpy1for Python tools and BLAST setupr_envfor DESeq2 + plotting heatmaps in R
conda activate plot-numpy11) Directory layout
From your project root:
.
├── CP052959.gb
├── MT880872.1.gb
├── results/star_salmon/degenes/
│ ├── Mitomycin_4h_vs_Untreated_4h-all_annotated.csv
│ ├── ...
└── subset_heatmaps/ # all scripts + outputs go hereCreate the output directory:
mkdir -p subset_heatmaps2) Step A/B/C gene set generation + batch intersection (one command)
This section generates:
geneset_A_phage_GeneID_plain.id(+GeneID.id)geneset_B_stress_GeneID_plain.id(+GeneID.id)geneset_C_biofilm_GeneID_plain.id(+GeneID.id)- plus all per-contrast
GOI_*files and*_matched.tsv
2.1 Script: extract CDS FASTA from MT880872.1.gb
Save as subset_heatmaps/extract_cds_fasta.py
#!/usr/bin/env python3
from Bio import SeqIO
import sys
gb = sys.argv[1]
out_fa = sys.argv[2]
rec = SeqIO.read(gb, "genbank")
with open(out_fa, "w") as out:
for f in rec.features:
if f.type != "CDS":
continue
locus = f.qualifiers.get("locus_tag", ["NA"])[0]
seq = f.extract(rec.seq)
out.write(f">{locus}\n{str(seq).upper()}\n")2.2 Script: BLAST hit mapping → CP052959 GeneID_plain set (geneset A)
Save as subset_heatmaps/blast_hits_to_geneset.py
#!/usr/bin/env python3
import sys
import pandas as pd
from Bio import SeqIO
blast6 = sys.argv[1]
cp_gb = sys.argv[2]
prefix = sys.argv[3] # e.g. subset_heatmaps/geneset_A_phage
# Load CP052959 CDS intervals
rec = SeqIO.read(cp_gb, "genbank")
cds = []
for f in rec.features:
if f.type != "CDS":
continue
locus = f.qualifiers.get("locus_tag", [None])[0]
if locus is None:
continue
start = int(f.location.start) + 1
end = int(f.location.end)
cds.append((locus, start, end))
cds_df = pd.DataFrame(cds, columns=["GeneID_plain","start","end"])
# Load BLAST tabular (outfmt 6)
cols = ["qseqid","sseqid","pident","length","mismatch","gapopen","qstart","qend",
"sstart","send","evalue","bitscore"]
b = pd.read_csv(blast6, sep="\t", names=cols)
# Normalize subject coordinates
b["smin"] = b[["sstart","send"]].min(axis=1)
b["smax"] = b[["sstart","send"]].max(axis=1)
# Filter for strong hits (tune if needed)
b = b[(b["pident"] >= 90.0) & (b["length"] >= 100)]
hits = set()
for _, r in b.iterrows():
ov = cds_df[(r["smin"] <= cds_df["end"]) & (r["smax"] >= cds_df["start"])]
hits.update(ov["GeneID_plain"].unique().tolist())
hits = sorted(hits)
plain_path = f"{prefix}_GeneID_plain.id"
geneid_path = f"{prefix}_GeneID.id"
pd.Series(hits).to_csv(plain_path, index=False, header=False)
pd.Series(["gene-" + x for x in hits]).to_csv(geneid_path, index=False, header=False)
print(f"Wrote {len(hits)} genes:")
print(" ", plain_path)
print(" ", geneid_path)2.3 Script: keyword-based genesets B/C from CP052959 annotations
Save as subset_heatmaps/geneset_by_keywords.py
#!/usr/bin/env python3
import sys, re
import pandas as pd
from Bio import SeqIO
cp_gb = sys.argv[1]
mode = sys.argv[2] # "stress" or "biofilm"
prefix = sys.argv[3] # e.g. subset_heatmaps/geneset_B_stress
rec = SeqIO.read(cp_gb, "genbank")
rows=[]
for f in rec.features:
if f.type != "CDS":
continue
locus = f.qualifiers.get("locus_tag", [None])[0]
if locus is None:
continue
gene = (f.qualifiers.get("gene", [""])[0] or "")
product = (f.qualifiers.get("product", [""])[0] or "")
note = "; ".join(f.qualifiers.get("note", [])) if f.qualifiers.get("note") else ""
text = " ".join([gene, product, note]).strip()
rows.append((locus, gene, product, note, text))
df = pd.DataFrame(rows, columns=["GeneID_plain","gene","product","note","text"])
if mode == "stress":
rgx = re.compile(
r"\b(stress|heat shock|chaperone|dnaK|groEL|groES|clp|thioredoxin|peroxiredoxin|catalase|superoxide|"
r"recA|lexA|uvr|mutS|mutL|usp|osm|sox|katA|sod)\b",
re.I
)
elif mode == "biofilm":
rgx = re.compile(
r"\b(biofilm|ica|pga|polysaccharide|PIA|adhesin|MSCRAMM|fibrinogen-binding|fibronectin-binding|"
r"clumping factor|sortase|autolysin|atl|nuclease|DNase|protease|dispersin|luxS|agr|sarA|dlt)\b",
re.I
)
else:
raise SystemExit("mode must be stress or biofilm")
sel = df[df["text"].apply(lambda x: bool(rgx.search(x)))].copy()
hits = sorted(sel["GeneID_plain"].unique())
plain_path = f"{prefix}_GeneID_plain.id"
geneid_path = f"{prefix}_GeneID.id"
sel_path = f"{prefix}_hits.tsv"
pd.Series(hits).to_csv(plain_path, index=False, header=False)
pd.Series(["gene-" + x for x in hits]).to_csv(geneid_path, index=False, header=False)
sel.drop(columns=["text"]).to_csv(sel_path, sep="\t", index=False)
print(f"{mode}: wrote {len(hits)} genes:")
print(" ", plain_path)
print(" ", geneid_path)
print(" ", sel_path)2.4 Script: intersect each DE table with A/B/C (no cutoff) and write GOI lists + matched TSV
Save as subset_heatmaps/make_goi_lists_batch.py
#!/usr/bin/env python3
import sys, glob, os
import pandas as pd
de_dir = sys.argv[1] # results/star_salmon/degenes
out_dir = sys.argv[2] # subset_heatmaps
genesetA_plain = sys.argv[3] # subset_heatmaps/geneset_A_phage_GeneID_plain.id
genesetB_plain = sys.argv[4] # subset_heatmaps/geneset_B_stress_GeneID_plain.id
genesetC_plain = sys.argv[5] # subset_heatmaps/geneset_C_biofilm_GeneID_plain.id
def load_plain_ids(path):
with open(path) as f:
return set(x.strip() for x in f if x.strip())
A = load_plain_ids(genesetA_plain)
B = load_plain_ids(genesetB_plain)
C = load_plain_ids(genesetC_plain)
def pick_id_cols(df):
geneid = "GeneID" if "GeneID" in df.columns else None
plain = "GeneID_plain" if "GeneID_plain" in df.columns else None
if plain is None and "GeneName" in df.columns:
plain = "GeneName"
return geneid, plain
os.makedirs(out_dir, exist_ok=True)
for csv in sorted(glob.glob(os.path.join(de_dir, "*-all_annotated.csv"))):
base = os.path.basename(csv).replace("-all_annotated.csv", "")
df = pd.read_csv(csv)
geneid_col, plain_col = pick_id_cols(df)
if plain_col is None:
raise SystemExit(f"Cannot find GeneID_plain/GeneName in {csv}")
df["__plain__"] = df[plain_col].astype(str).str.replace("^gene-","", regex=True)
def write_set(tag, S):
sub = df[df["__plain__"].isin(S)].copy()
out_plain = os.path.join(out_dir, f"GOI_{base}_{tag}_GeneID_plain.id")
out_geneid = os.path.join(out_dir, f"GOI_{base}_{tag}_GeneID.id")
out_tsv = os.path.join(out_dir, f"{base}_{tag}_matched.tsv")
sub["__plain__"].drop_duplicates().to_csv(out_plain, index=False, header=False)
pd.Series(["gene-"+x for x in sub["__plain__"].drop_duplicates()]).to_csv(out_geneid, index=False, header=False)
sub.to_csv(out_tsv, sep="\t", index=False)
print(f"{base} {tag}: {sub.shape[0]} rows, {sub['__plain__'].nunique()} genes")
write_set("A_phage", A)
write_set("B_stress", B)
write_set("C_biofilm", C)2.5 Driver: run everything with one command
Save as subset_heatmaps/run_subset_setup.sh
#!/usr/bin/env bash
set -euo pipefail
DE_DIR="./results/star_salmon/degenes"
OUT_DIR="./subset_heatmaps"
CP_GB="CP052959.gb"
PHAGE_GB="MT880872.1.gb"
mkdir -p "$OUT_DIR"
echo "[INFO] Using DE_DIR=$DE_DIR"
ls -lh "$DE_DIR"/*-all_annotated.csv
# ---- A) BLAST-based phage/prophage geneset ----
python - <<'PY'
from Bio import SeqIO
rec=SeqIO.read("CP052959.gb","genbank")
SeqIO.write(rec, "subset_heatmaps/CP052959.fna", "fasta")
PY
python subset_heatmaps/extract_cds_fasta.py "$PHAGE_GB" "$OUT_DIR/MT880872_CDS.fna"
makeblastdb -in "$OUT_DIR/CP052959.fna" -dbtype nucl -out "$OUT_DIR/CP052959_db" >/dev/null
blastn \
-query "$OUT_DIR/MT880872_CDS.fna" \
-db "$OUT_DIR/CP052959_db" \
-out "$OUT_DIR/MT_vs_CP.blast6" \
-outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore" \
-evalue 1e-10
python subset_heatmaps/blast_hits_to_geneset.py \
"$OUT_DIR/MT_vs_CP.blast6" "$CP_GB" "$OUT_DIR/geneset_A_phage"
# ---- B/C) keyword-based genesets ----
python subset_heatmaps/geneset_by_keywords.py "$CP_GB" stress "$OUT_DIR/geneset_B_stress"
python subset_heatmaps/geneset_by_keywords.py "$CP_GB" biofilm "$OUT_DIR/geneset_C_biofilm"
# ---- Batch: intersect each DE CSV with the genesets (no cutoff) ----
python subset_heatmaps/make_goi_lists_batch.py \
"$DE_DIR" "$OUT_DIR" \
"$OUT_DIR/geneset_A_phage_GeneID_plain.id" \
"$OUT_DIR/geneset_B_stress_GeneID_plain.id" \
"$OUT_DIR/geneset_C_biofilm_GeneID_plain.id"
echo "[INFO] Done. GOI lists are in $OUT_DIR"
ls -1 "$OUT_DIR"/GOI_*_GeneID.id | headRun it:
bash subset_heatmaps/run_subset_setup.shAt this point you will have all *_matched.tsv files required for plotting, e.g.:
Mitomycin_4h_vs_Untreated_4h_A_phage_matched.tsvMoxi_4h_vs_Untreated_4h_A_phage_matched.tsv- … (for 8h/18h and B/C)
3) No-cutoff heatmaps (merged Untreated + Mitomycin + Moxi → 9 figures)
Now switch to your R environment and build the rlog (rld) expression matrix from Salmon quantifications.
conda activate r_env3.1 Build rld from Salmon outputs (R)
library(tximport)
library(DESeq2)
setwd("~/DATA/Data_JuliaFuchs_RNAseq_2025/results/star_salmon")
files <- c(
"Untreated_4h_r1" = "./Untreated_4h_1a/quant.sf",
"Untreated_4h_r2" = "./Untreated_4h_1b/quant.sf",
"Untreated_4h_r3" = "./Untreated_4h_1c/quant.sf",
"Untreated_8h_r1" = "./Untreated_8h_1d/quant.sf",
"Untreated_8h_r2" = "./Untreated_8h_1e/quant.sf",
"Untreated_8h_r3" = "./Untreated_8h_1f/quant.sf",
"Untreated_18h_r1" = "./Untreated_18h_1g/quant.sf",
"Untreated_18h_r2" = "./Untreated_18h_1h/quant.sf",
"Untreated_18h_r3" = "./Untreated_18h_1i/quant.sf",
"Mitomycin_4h_r1" = "./Mitomycin_4h_2a/quant.sf",
"Mitomycin_4h_r2" = "./Mitomycin_4h_2b/quant.sf",
"Mitomycin_4h_r3" = "./Mitomycin_4h_2c/quant.sf",
"Mitomycin_8h_r1" = "./Mitomycin_8h_2d/quant.sf",
"Mitomycin_8h_r2" = "./Mitomycin_8h_2e/quant.sf",
"Mitomycin_8h_r3" = "./Mitomycin_8h_2f/quant.sf",
"Mitomycin_18h_r1" = "./Mitomycin_18h_2g/quant.sf",
"Mitomycin_18h_r2" = "./Mitomycin_18h_2h/quant.sf",
"Mitomycin_18h_r3" = "./Mitomycin_18h_2i/quant.sf",
"Moxi_4h_r1" = "./Moxi_4h_3a/quant.sf",
"Moxi_4h_r2" = "./Moxi_4h_3b/quant.sf",
"Moxi_4h_r3" = "./Moxi_4h_3c/quant.sf",
"Moxi_8h_r1" = "./Moxi_8h_3d/quant.sf",
"Moxi_8h_r2" = "./Moxi_8h_3e/quant.sf",
"Moxi_8h_r3" = "./Moxi_8h_3f/quant.sf",
"Moxi_18h_r1" = "./Moxi_18h_3g/quant.sf",
"Moxi_18h_r2" = "./Moxi_18h_3h/quant.sf",
"Moxi_18h_r3" = "./Moxi_18h_3i/quant.sf"
)
txi <- tximport(files, type = "salmon", txIn = TRUE, txOut = TRUE)
replicate <- factor(rep(c("r1","r2","r3"), 9))
condition <- factor(c(
rep("Untreated_4h",3), rep("Untreated_8h",3), rep("Untreated_18h",3),
rep("Mitomycin_4h",3), rep("Mitomycin_8h",3), rep("Mitomycin_18h",3),
rep("Moxi_4h",3), rep("Moxi_8h",3), rep("Moxi_18h",3)
))
colData <- data.frame(condition=condition, replicate=replicate, row.names=names(files))
dds <- DESeqDataSetFromTximport(txi, colData, design = ~ condition)
rld <- rlogTransformation(dds)3.2 Plot merged 3-condition subset heatmaps (R)
suppressPackageStartupMessages(library(gplots))
need <- c("openxlsx")
to_install <- setdiff(need, rownames(installed.packages()))
if (length(to_install)) install.packages(to_install, repos = "https://cloud.r-project.org")
suppressPackageStartupMessages(library(openxlsx))
in_dir <- "subset_heatmaps"
out_dir <- file.path(in_dir, "heatmaps_merged3")
dir.create(out_dir, showWarnings = FALSE, recursive = TRUE)
pick_col <- function(df, candidates) {
hit <- intersect(candidates, names(df))
if (length(hit) == 0) return(NA_character_)
hit[1]
}
strip_gene_prefix <- function(x) sub("^gene[-_]", "", x)
match_tags <- function(nms, tags) {
pat <- paste0("(^|_)(?:", paste(tags, collapse = "|"), ")(_|$)")
grepl(pat, nms, perl = TRUE)
}
detect_tag <- function(nm, tags) {
hits <- vapply(tags, function(t)
grepl(paste0("(^|_)", t, "(_|$)"), nm, perl = TRUE), logical(1))
if (!any(hits)) NA_character_ else tags[which(hits)[1]]
}
make_pretty_labels <- function(gene_ids_in_matrix, id2name, id2desc) {
plain <- strip_gene_prefix(gene_ids_in_matrix)
nm <- unname(id2name[plain]); ds <- unname(id2desc[plain])
nm[is.na(nm)] <- ""; ds[is.na(ds)] <- ""
nm2 <- ifelse(nzchar(nm), nm, plain)
lbl <- ifelse(nzchar(ds), paste0(nm2, " (", ds, ")"), nm2)
make.unique(lbl, sep = "_")
}
if (exists("rld")) {
expr_all <- assay(rld)
} else if (exists("vsd")) {
expr_all <- assay(vsd)
} else {
stop("Neither 'rld' nor 'vsd' exists. Create/load it before running this script.")
}
expr_all <- as.matrix(expr_all)
mat_ids <- rownames(expr_all)
if (is.null(mat_ids)) stop("Expression matrix has no rownames.")
times <- c("4h", "8h", "18h")
tags <- c("A_phage", "B_stress", "C_biofilm")
cond_order_template <- c("Untreated_%s", "Mitomycin_%s", "Moxi_%s")
for (tt in times) {
for (tag in tags) {
f_mito <- file.path(in_dir, sprintf("Mitomycin_%s_vs_Untreated_%s_%s_matched.tsv", tt, tt, tag))
f_moxi <- file.path(in_dir, sprintf("Moxi_%s_vs_Untreated_%s_%s_matched.tsv", tt, tt, tag))
if (!file.exists(f_mito) || !file.exists(f_moxi)) next
df1 <- read.delim(f_mito, sep = "\t", header = TRUE, stringsAsFactors = FALSE, check.names = FALSE)
df2 <- read.delim(f_moxi, sep = "\t", header = TRUE, stringsAsFactors = FALSE, check.names = FALSE)
id_col_1 <- pick_col(df1, c("GeneID","GeneID_plain","Gene_Id","gene_id","locus_tag","LocusTag","ID"))
id_col_2 <- pick_col(df2, c("GeneID","GeneID_plain","Gene_Id","gene_id","locus_tag","LocusTag","ID"))
if (is.na(id_col_1) || is.na(id_col_2)) next
name_col_1 <- pick_col(df1, c("GeneName","Preferred_name","gene","Symbol","Name"))
name_col_2 <- pick_col(df2, c("GeneName","Preferred_name","gene","Symbol","Name"))
desc_col_1 <- pick_col(df1, c("Description","product","Product","annotation","Annot","note"))
desc_col_2 <- pick_col(df2, c("Description","product","Product","annotation","Annot","note"))
g1 <- unique(trimws(df1[[id_col_1]])); g1 <- g1[nzchar(g1)]
g2 <- unique(trimws(df2[[id_col_2]])); g2 <- g2[nzchar(g2)]
GOI_raw <- unique(c(g1, g2))
present <- intersect(mat_ids, GOI_raw)
if (!length(present)) {
present <- unique(mat_ids[strip_gene_prefix(mat_ids) %in% strip_gene_prefix(GOI_raw)])
}
if (!length(present)) next
getcol <- function(df, col, n) if (is.na(col)) rep("", n) else as.character(df[[col]])
plain1 <- strip_gene_prefix(as.character(df1[[id_col_1]]))
plain2 <- strip_gene_prefix(as.character(df2[[id_col_2]]))
nm1 <- getcol(df1, name_col_1, nrow(df1)); nm2 <- getcol(df2, name_col_2, nrow(df2))
ds1 <- getcol(df1, desc_col_1, nrow(df1)); ds2 <- getcol(df2, desc_col_2, nrow(df2))
nm1[is.na(nm1)] <- ""; nm2[is.na(nm2)] <- ""
ds1[is.na(ds1)] <- ""; ds2[is.na(ds2)] <- ""
keys_all <- unique(c(plain1, plain2))
id2name <- setNames(rep("", length(keys_all)), keys_all)
id2desc <- setNames(rep("", length(keys_all)), keys_all)
fill_map <- function(keys, vals, mp) {
for (i in seq_along(keys)) {
k <- keys[i]; v <- vals[i]
if (!nzchar(k)) next
if (!nzchar(mp[[k]]) && nzchar(v)) mp[[k]] <- v
}
mp
}
id2name <- fill_map(plain1, nm1, id2name); id2name <- fill_map(plain2, nm2, id2name)
id2desc <- fill_map(plain1, ds1, id2desc); id2desc <- fill_map(plain2, ds2, id2desc)
cond_tags <- sprintf(cond_order_template, tt)
keep_cols <- match_tags(colnames(expr_all), cond_tags)
if (!any(keep_cols)) next
sub_idx <- which(keep_cols)
sub_names <- colnames(expr_all)[sub_idx]
cond_for_col <- vapply(sub_names, detect_tag, character(1), tags = cond_tags)
cond_rank <- match(cond_for_col, cond_tags)
ord <- order(cond_rank, sub_names)
sub_idx <- sub_idx[ord]
expr_sub <- expr_all[present, sub_idx, drop = FALSE]
row_ok <- apply(expr_sub, 1, function(x) is.finite(sum(x)) && var(x, na.rm = TRUE) > 0)
datamat <- expr_sub[row_ok, , drop = FALSE]
if (nrow(datamat) < 2) next
hr <- hclust(as.dist(1 - cor(t(datamat), method = "pearson")), method = "complete")
mycl <- cutree(hr, h = max(hr$height) / 1.1)
palette_base <- c("yellow","blue","orange","magenta","cyan","red","green","maroon",
"lightblue","pink","purple","lightcyan","salmon","lightgreen")
mycol <- palette_base[(as.vector(mycl) - 1) %% length(palette_base) + 1]
labRow <- make_pretty_labels(rownames(datamat), id2name, id2desc)
labCol <- gsub("_", " ", colnames(datamat))
gene_id <- rownames(datamat)
gene_plain <- strip_gene_prefix(gene_id)
gene_name <- unname(id2name[gene_plain]); gene_name[is.na(gene_name)] <- ""
gene_desc <- unname(id2desc[gene_plain]); gene_desc[is.na(gene_desc)] <- ""
out_tbl <- data.frame(
GeneID = gene_id,
GeneID_plain = gene_plain,
GeneName = ifelse(nzchar(gene_name), gene_name, gene_plain),
Description = gene_desc,
datamat,
check.names = FALSE,
stringsAsFactors = FALSE
)
base <- sprintf("%s_%s_merged3", tt, tag)
out_xlsx <- file.path(out_dir, paste0("table_", base, ".xlsx"))
write.xlsx(out_tbl, out_xlsx, overwrite = TRUE)
out_png <- file.path(out_dir, paste0("heatmap_", base, ".png"))
cex_row <- if (nrow(datamat) > 600) 0.90 else if (nrow(datamat) > 300) 1.05 else 1.30
height <- max(1600, min(18000, 34 * nrow(datamat)))
png(out_png, width = 2200, height = height)
heatmap.2(
datamat,
Rowv = as.dendrogram(hr),
Colv = FALSE,
dendrogram = "row",
col = bluered(75),
scale = "row",
trace = "none",
density.info = "none",
RowSideColors = mycol,
margins = c(12, 60),
labRow = labRow,
labCol = labCol,
cexRow = cex_row,
cexCol = 2.0,
srtCol = 15,
key = FALSE
)
dev.off()
message("WROTE: ", out_png)
message("WROTE: ", out_xlsx)
}
}
message("Done. Output dir: ", out_dir)Run it:
setwd("~/DATA/Data_JuliaFuchs_RNAseq_2025")
source("subset_heatmaps/draw_9_merged_heatmaps.R")3.3 Optional: Plot 2-condition subset heatmaps (R)
#!/usr/bin/env Rscript
## =============================================================
## Draw 18 subset heatmaps using *_matched.tsv as input
## Output: subset_heatmaps/heatmaps_from_matched/
##
## Requirements:
## - rld or vsd exists in environment (DESeq2 transform)
## If running as Rscript, you must load/create rld/vsd BEFORE sourcing this file
## (see the note at the bottom for the "source()" way)
##
## Matched TSV must contain GeneID or GeneID_plain (or GeneName) columns.
## =============================================================
suppressPackageStartupMessages(library(gplots))
in_dir <- "subset_heatmaps"
out_dir <- file.path(in_dir, "heatmaps_from_matched")
dir.create(out_dir, showWarnings = FALSE, recursive = TRUE)
# -------------------------
# Helper functions
# -------------------------
pick_col <- function(df, candidates) {
hit <- intersect(candidates, names(df))
if (length(hit) == 0) return(NA_character_)
hit[1]
}
strip_gene_prefix <- function(x) sub("^gene[-_]", "", x)
split_contrast_groups <- function(x) {
parts <- strsplit(x, "_vs_", fixed = TRUE)[[1]]
if (length(parts) != 2L) stop("Contrast must be in form A_vs_B: ", x)
parts
}
match_tags <- function(nms, tags) {
pat <- paste0("(^|_)(?:", paste(tags, collapse = "|"), ")(_|$)")
grepl(pat, nms, perl = TRUE)
}
# -------------------------
# Get expression matrix
# -------------------------
if (exists("rld")) {
expr_all <- assay(rld)
} else if (exists("vsd")) {
expr_all <- assay(vsd)
} else {
stop("Neither 'rld' nor 'vsd' exists. Create/load it before running this script.")
}
expr_all <- as.matrix(expr_all)
mat_ids <- rownames(expr_all)
if (is.null(mat_ids)) stop("Expression matrix has no rownames.")
# -------------------------
# List your 18 matched inputs
# -------------------------
matched_files <- c(
"Mitomycin_4h_vs_Untreated_4h_A_phage_matched.tsv",
"Mitomycin_4h_vs_Untreated_4h_B_stress_matched.tsv",
"Mitomycin_4h_vs_Untreated_4h_C_biofilm_matched.tsv",
"Mitomycin_8h_vs_Untreated_8h_A_phage_matched.tsv",
"Mitomycin_8h_vs_Untreated_8h_B_stress_matched.tsv",
"Mitomycin_8h_vs_Untreated_8h_C_biofilm_matched.tsv",
"Mitomycin_18h_vs_Untreated_18h_A_phage_matched.tsv",
"Mitomycin_18h_vs_Untreated_18h_B_stress_matched.tsv",
"Mitomycin_18h_vs_Untreated_18h_C_biofilm_matched.tsv",
"Moxi_4h_vs_Untreated_4h_A_phage_matched.tsv",
"Moxi_4h_vs_Untreated_4h_B_stress_matched.tsv",
"Moxi_4h_vs_Untreated_4h_C_biofilm_matched.tsv",
"Moxi_8h_vs_Untreated_8h_A_phage_matched.tsv",
"Moxi_8h_vs_Untreated_8h_B_stress_matched.tsv",
"Moxi_8h_vs_Untreated_8h_C_biofilm_matched.tsv",
"Moxi_18h_vs_Untreated_18h_A_phage_matched.tsv",
"Moxi_18h_vs_Untreated_18h_B_stress_matched.tsv",
"Moxi_18h_vs_Untreated_18h_C_biofilm_matched.tsv"
)
matched_paths <- file.path(in_dir, matched_files)
# -------------------------
# Main loop
# -------------------------
for (path in matched_paths) {
if (!file.exists(path)) {
message("SKIP missing: ", path)
next
}
base <- sub("_matched\\.tsv$", "", basename(path))
# base looks like: Mitomycin_4h_vs_Untreated_4h_A_phage
# split base into contrast + tag (last 2 underscore fields are the tag)
parts <- strsplit(base, "_")[[1]]
if (length(parts) < 6) {
message("SKIP unexpected name: ", base)
next
}
# infer tag as last 2 parts: e.g. A_phage / B_stress / C_biofilm
tag <- paste0(parts[length(parts)-1], "_", parts[length(parts)])
# contrast is the rest
contrast <- paste(parts[1:(length(parts)-2)], collapse = "_")
# read matched TSV
df <- read.delim(path, sep = "\t", header = TRUE, stringsAsFactors = FALSE, check.names = FALSE)
id_col <- pick_col(df, c("GeneID", "GeneID_plain", "GeneName", "Gene_Id", "gene_id", "locus_tag", "LocusTag", "ID"))
if (is.na(id_col)) {
message("SKIP (no ID col): ", path)
next
}
GOI_raw <- unique(trimws(df[[id_col]]))
GOI_raw <- GOI_raw[nzchar(GOI_raw)]
# match GOI to matrix ids robustly
present <- intersect(mat_ids, GOI_raw)
if (!length(present)) {
present <- unique(mat_ids[strip_gene_prefix(mat_ids) %in% strip_gene_prefix(GOI_raw)])
}
if (!length(present)) {
message("SKIP (no GOI matched matrix): ", base)
next
}
# subset columns for the two groups
groups <- split_contrast_groups(contrast)
keep_cols <- match_tags(colnames(expr_all), groups)
if (!any(keep_cols)) {
message("SKIP (no columns matched groups): ", contrast)
next
}
cols_idx <- which(keep_cols)
sub_colnames <- colnames(expr_all)[cols_idx]
# put Untreated first (2nd group in "Treated_vs_Untreated")
ord <- order(!grepl(paste0("(^|_)", groups[2], "(_|$)"), sub_colnames, perl = TRUE))
cols_idx <- cols_idx[ord]
expr_sub <- expr_all[present, cols_idx, drop = FALSE]
# remove constant/NA rows
row_ok <- apply(expr_sub, 1, function(x) is.finite(sum(x)) && var(x, na.rm = TRUE) > 0)
datamat <- expr_sub[row_ok, , drop = FALSE]
if (nrow(datamat) < 2) {
message("SKIP (too few rows after filtering): ", base)
next
}
# clustering
hr <- hclust(as.dist(1 - cor(t(datamat), method = "pearson")), method = "complete")
mycl <- cutree(hr, h = max(hr$height) / 1.1)
palette_base <- c("yellow","blue","orange","magenta","cyan","red","green","maroon",
"lightblue","pink","purple","lightcyan","salmon","lightgreen")
mycol <- palette_base[(as.vector(mycl) - 1) %% length(palette_base) + 1]
# labels
labRow <- rownames(datamat)
labRow <- sub("^gene-", "", labRow)
labRow <- sub("^rna-", "", labRow)
labCol <- colnames(datamat)
labCol <- gsub("_", " ", labCol)
# output sizes
height <- max(900, min(12000, 25 * nrow(datamat)))
out_png <- file.path(out_dir, paste0("heatmap_", base, ".png"))
out_mat <- file.path(out_dir, paste0("matrix_", base, ".csv"))
write.csv(as.data.frame(datamat), out_mat, quote = FALSE)
png(out_png, width = 1100, height = height)
heatmap.2(
datamat,
Rowv = as.dendrogram(hr),
Colv = FALSE,
dendrogram = "row",
col = bluered(75),
scale = "row",
trace = "none",
density.info = "none",
RowSideColors = mycol,
margins = c(10, 15),
sepwidth = c(0, 0),
labRow = labRow,
labCol = labCol,
cexRow = if (nrow(datamat) > 500) 0.6 else 1.0,
cexCol = 1.7,
srtCol = 15,
lhei = c(0.01, 4),
lwid = c(0.5, 4),
key = FALSE
)
dev.off()
message("WROTE: ", out_png)
}
message("All done. Output dir: ", out_dir)Run it:
setwd("~/DATA/Data_JuliaFuchs_RNAseq_2025")
source("subset_heatmaps/draw_18_heatmaps_from_matched.R")4) Optional: Update README_Heatmap to support “GOI file OR no-cutoff”
If you still use the older README_Heatmap logic that expects *-up.id and *-down.id, replace the GOI-building block with this (single GOI list or whole CSV with no cutoff):
geneset_file <- NA_character_ # e.g. "subset_heatmaps/GOI_Mitomycin_4h_vs_Untreated_4h_A_phage_GeneID.id"
use_all_genes_no_cutoff <- FALSE
if (!is.na(geneset_file) && file.exists(geneset_file)) {
GOI <- read_ids_from_file(geneset_file)
} else if (isTRUE(use_all_genes_no_cutoff)) {
all_path <- file.path("./results/star_salmon/degenes", paste0(contrast, "-all_annotated.csv"))
ann <- read.csv(all_path, stringsAsFactors = FALSE, check.names = FALSE)
id_col <- if ("GeneID" %in% names(ann)) "GeneID" else if ("GeneID_plain" %in% names(ann)) "GeneID_plain" else NA_character_
if (is.na(id_col)) stop("No GeneID / GeneID_plain in: ", all_path)
GOI <- unique(trimws(gsub('"', "", ann[[id_col]])))
GOI <- GOI[nzchar(GOI)]
} else {
stop("Set geneset_file OR set use_all_genes_no_cutoff <- TRUE")
}
present <- intersect(rownames(RNASeq.NoCellLine), GOI)
if (!length(present)) stop("None of the GOI found in expression matrix rownames.")
GOI <- present5) Script inventory (bash + python)
Bash
subset_heatmaps/run_subset_setup.sh
Python
subset_heatmaps/extract_cds_fasta.pysubset_heatmaps/blast_hits_to_geneset.pysubset_heatmaps/geneset_by_keywords.pysubset_heatmaps/make_goi_lists_batch.py
R
subset_heatmaps/draw_9_merged_heatmaps.Rsubset_heatmaps/draw_18_heatmaps_from_matched.R
This post is a lab wiki / GitHub README / methods note, every script referenced is included in full above (and can be copied into subset_heatmaps/ directly).