-
Input data
WaGa wt cells (nf774* (Considering to be deleted, due to possibly be an outlier, but in the current version, it is still included in the analysis), nf961, nf962) WaGa wt_EV_RNA (nf657* (The sample was EXCLUDED, since it is obviously a outlier, not clustered with the other 2 samples), nf930, nf935) WaGa_sT_DMSO_EV_RNA (nf931, nf936, nf971) WaGa_sT_Dox_EV_RNA (nf932, nf937, nf972) WaGa_scr_DMSO_EV_RNA (nf933, nf938, nf973) WaGa_scr_Dox_EV_RNA (nf934, nf939, nf974) # --> In total, 17 samples MKL-1 wt cells (nf780*, nf796*, nf797*) MKL-1 wt_EV_RNA (nf655* (The sample was EXCLUDED), 2404, 2608) MKL-1_sT_DMSO_EV_RNA (2608, 2701, 2802) MKL-1_sT_Dox_EV_RNA (2608, 2701, 2802) MKL-1_scr_DMSO_EV_RNA (2608, 2701, 2802) MKL-1_scr_Dox_EV_RNA (2608, 2701, 2802) # --> In total, 18 samples #Note that the real paths are as follows: #./20260506_AV243904_0073_A/2404_MKL1_wt_EVs/2404_MKL1_wt_EVs_R1.fastq.gz, ./20260506_AV243904_0073_A/2608_MKL1_wt_EVs/2608_MKL1_wt_EVs_R1.fastq.gz #./20260506_AV243904_0073_A/2608_MKL1_sT_DMSO/2608_MKL1_sT_DMSO_R1.fastq.gz, ./20260506_AV243904_0073_A/2701_MKL1_sT_DMSO/2701_MKL1_sT_DMSO_R1.fastq.gz, ./20260506_AV243904_0073_A/2802_MKL1_sT_DMSO/2802_MKL1_sT_DMSO_R1.fastq.gz #./20260506_AV243904_0073_A/2608_MKL1_sT_Dox/2608_MKL1_sT_Dox_R1.fastq.gz, ./20260506_AV243904_0073_A/2701_MKL1_sT_Dox/2701_MKL1_sT_Dox_R1.fastq.gz, ./20260506_AV243904_0073_A/2802_MKL1_sT_Dox/2802_MKL1_sT_Dox_R1.fastq.gz #./20260506_AV243904_0073_A/2608_MKL1_scr_DMSO/2608_MKL1_scr_DMSO_R1.fastq.gz, ./20260506_AV243904_0073_A/2701_MKL1_scr_DMSO/2701_MKL1_scr_DMSO_R1.fastq.gz, ./20260506_AV243904_0073_A/2802_MKL1_scr_DMSO/2802_MKL1_scr_DMSO_R1.fastq.gz #./20260506_AV243904_0073_A/2608_MKL1_scr_Dox/2608_MKL1_scr_Dox_R1.fastq.gz, ./20260506_AV243904_0073_A/2701_MKL1_scr_Dox/2701_MKL1_scr_Dox_R1.fastq.gz, ./20260506_AV243904_0073_A/2802_MKL1_scr_Dox/2802_MKL1_scr_Dox_R1.fastq.gz -
Adapter trimming
#some common adapter sequences from different kits for reference: # - TruSeq Small RNA (Illumina): TGGAATTCTCGGGTGCCAAGG # - Small RNA Kits V1 (Illumina): TCGTATGCCGTCTTCTGCTTGT # - Small RNA Kits V1.5 (Illumina): ATCTCGTATGCCGTCTTCTGCTTG # - NEXTflex Small RNA Sequencing Kit v3 for Illumina Platforms (Bioo Scientific): TGGAATTCTCGGGTGCCAAGG # - LEXOGEN Small RNA-Seq Library Prep Kit (Illumina): TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC * mkdir Data_Ute_smallRNA_via_exceRpt_workspace/trimmed; cd Data_Ute_smallRNA_via_exceRpt_workspace/trimmed echo "------------------------------------ cutadapting nf774 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf774.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf774/0403_WaGa_wt_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf657 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf657.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti/2021_nf_ute_smallRNA/nf657/WaGa_derived_EV_miRNA_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf655 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf655.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/210817_NB501882_0294_AHW5Y2BGXJ_smallRNA_Ute_newDemulti/2021_nf_ute_smallRNA/nf655/MKL_1_derived_EV_miRNA_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf780 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf780.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/220617_NB501882_0371_AH7572BGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf780/0505_MKL1_wt_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf796 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf796.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf796/MKL-1_wt_1_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf797 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf797.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_4/230623_newDemulti_smallRNAs/221216_NB501882_0404_AHLVNMBGXM_smallRNA_Ute_newDemulti/2022_nf_ute_smallRNA/nf797/MKL-1_wt_2_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf930 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf930.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf930/01_0505_WaGa_wt_EV_RNA_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf931 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf931.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf931/02_0505_WaGa_sT_DMSO_EV_RNA_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf932 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf932.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf932/03_0505_WaGa_sT_Dox_EV_RNA_S3_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf933 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf933.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf933/04_0505_WaGa_scr_DMSO_EV_RNA_S4_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf934 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf934.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf934/05_0505_WaGa_scr_Dox_EV_RNA_S5_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf935 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf935.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf935/06_1905_WaGa_wt_EV_RNA_S6_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf936 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf936.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf936/07_1905_WaGa_sT_DMSO_EV_RNA_S7_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf937 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf937.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf937/08_1905_WaGa_sT_Dox_EV_RNA_S8_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf938 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf938.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf938/09_1905_WaGa_scr_DMSO_EV_RNA_S9_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf939 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf939.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf939/10_1905_WaGa_scr_Dox_EV_RNA_S10_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf940 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf940.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf940/11_control_MKL1_S11_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf941 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf941.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/231016_NB501882_0435_AHG7HMBGXV/nf941/12_control_WaGa_S12_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf961 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf961.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf961/WaGaWTcells_1_S1_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf962 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf962.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf962/WaGaWTcells_2_S2_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf971 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf971.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf971/2001_WaGa_sT_DMSO_S3_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf972 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf972.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf972/2001_WaGa_sT_Dox_S4_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf973 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf973.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf973/2001_WaGa_scr_DMSO_S5_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting nf974 -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o nf974.fastq.gz ~/DATA/Data_Ute/Data_Ute_smallRNA_7/250411_VH00358_135_AAGKGLHM5/nf974/2001_WaGa_scr_Dox_S6_R1_001.fastq.gz >> LOG echo "------------------------------------ cutadapting 2404_MKL1_wt_EVs -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2404_MKL1_wt_EVs.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2404_MKL1_wt_EVs/2404_MKL1_wt_EVs_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2608_MKL1_wt_EVs -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2608_MKL1_wt_EVs.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2608_MKL1_wt_EVs/2608_MKL1_wt_EVs_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2608_MKL1_sT_DMSO -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2608_MKL1_sT_DMSO.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2608_MKL1_sT_DMSO/2608_MKL1_sT_DMSO_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2701_MKL1_sT_DMSO -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2701_MKL1_sT_DMSO.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2701_MKL1_sT_DMSO/2701_MKL1_sT_DMSO_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2802_MKL1_sT_DMSO -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2802_MKL1_sT_DMSO.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2802_MKL1_sT_DMSO/2802_MKL1_sT_DMSO_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2608_MKL1_sT_Dox -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2608_MKL1_sT_Dox.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2608_MKL1_sT_Dox/2608_MKL1_sT_Dox_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2701_MKL1_sT_Dox -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2701_MKL1_sT_Dox.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2701_MKL1_sT_Dox/2701_MKL1_sT_Dox_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2802_MKL1_sT_Dox -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2802_MKL1_sT_Dox.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2802_MKL1_sT_Dox/2802_MKL1_sT_Dox_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2608_MKL1_scr_DMSO -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2608_MKL1_scr_DMSO.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2608_MKL1_scr_DMSO/2608_MKL1_scr_DMSO_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2701_MKL1_scr_DMSO -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2701_MKL1_scr_DMSO.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2701_MKL1_scr_DMSO/2701_MKL1_scr_DMSO_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2802_MKL1_scr_DMSO -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2802_MKL1_scr_DMSO.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2802_MKL1_scr_DMSO/2802_MKL1_scr_DMSO_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2608_MKL1_scr_Dox -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2608_MKL1_scr_Dox.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2608_MKL1_scr_Dox/2608_MKL1_scr_Dox_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2701_MKL1_scr_Dox -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2701_MKL1_scr_Dox.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2701_MKL1_scr_Dox/2701_MKL1_scr_Dox_R1.fastq.gz >> LOG echo "------------------------------------ cutadapting 2802_MKL1_scr_Dox -----------------------------------" >> LOG cutadapt -a TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC -q 20 --minimum-length 5 --trim-n -o 2802_MKL1_scr_Dox.fastq.gz ~/DATA/Data_Ute_smallRNA/20260506_AV243904_0073_A/2802_MKL1_scr_Dox/2802_MKL1_scr_Dox_R1.fastq.gz >> LOG -
Install exceRpt (https://github.gersteinlab.org/exceRpt/)
docker pull rkitchen/excerpt mkdir MyexceRptDatabase cd /mnt/nvme0n1p1/MyexceRptDatabase wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg38_lowmem.tgz tar -xvf exceRptDB_v4_hg38_lowmem.tgz #http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg19_lowmem.tgz #http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg38_lowmem.tgz #http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_mm10_lowmem.tgz wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_EXOmiRNArRNA.tgz tar -xvf exceRptDB_v4_EXOmiRNArRNA.tgz wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_EXOGenomes.tgz tar -xvf exceRptDB_v4_EXOGenomes.tgz # List extracted hg38 directory structure find hg38 -type f | sed 's|^hg38/||' | sort > extracted_hg38.txt comm -3 extracted_hg38.txt <(tar -tf exceRptDB_v4_hg38_lowmem.tgz | grep '^hg38/' | sed 's|^hg38/||' | sort) # --> DIR hg38 tar -tf exceRptDB_v4_EXOmiRNArRNA.tgz # --> DIR ribosomeDatabase, NCBI_taxonomy_taxdump, miRBase tar -tf exceRptDB_v4_EXOGenomes.tgz # --> Genomes_BacteriaFungiMammalPlantProtistVirus -
Run exceRpt
#[---- REAL_RUNNING_COMPLETE_DB ---->] #NOTE that if not renamed in the input files, then have to RENAME all files recursively by removing "_cutadapted.fastq" in all names in _CORE_RESULTS_v4.6.3.tgz (first unzip, removing, then zip, mv to ../results_g). cd trimmed for file in *.fastq.gz; do echo "mv \"$file\" \"${file/.fastq/}\"" done mkdir results for sample in nf780 nf796 nf797 nf655 nf774 nf961 nf962 nf657 nf930 nf935 nf931 nf936 nf971 nf932 nf937 nf972 nf933 nf938 nf973 nf934 nf939 nf974; do docker run -v ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/trimmed:/exceRptInput \ -v ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/results:/exceRptOutput \ -v /mnt/nvme0n1p1/MyexceRptDatabase:/exceRpt_DB \ -t rkitchen/excerpt \ INPUT_FILE_PATH=/exceRptInput/${sample}.gz MAIN_ORGANISM_GENOME_ID=hg38 N_THREADS=50 JAVA_RAM='200G' MAP_EXOGENOUS=on done for sample in 2404_MKL1_wt_EVs 2608_MKL1_wt_EVs 2608_MKL1_sT_DMSO 2701_MKL1_sT_DMSO 2802_MKL1_sT_DMSO 2608_MKL1_sT_Dox 2701_MKL1_sT_Dox 2802_MKL1_sT_Dox 2608_MKL1_scr_DMSO 2701_MKL1_scr_DMSO 2802_MKL1_scr_DMSO 2608_MKL1_scr_Dox 2701_MKL1_scr_Dox 2802_MKL1_scr_Dox; do docker run -v ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/trimmed:/exceRptInput \ -v ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/results:/exceRptOutput \ -v /mnt/nvme3n1p1/MyexceRptDatabase:/exceRpt_DB \ -t rkitchen/excerpt \ INPUT_FILE_PATH=/exceRptInput/${sample}.gz MAIN_ORGANISM_GENOME_ID=hg38 N_THREADS=50 JAVA_RAM='200G' MAP_EXOGENOUS=on done #DEBUG the excerpt env docker inspect rkitchen/excerpt:latest # Without /bin/bash → May run and exit immediately #docker run -it rkitchen/excerpt # With /bin/bash → Stays open for interaction docker run -it --entrypoint /bin/bash rkitchen/excerpt -
Processing exceRpt output from multiple samples
cd ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/exceRpt-master mamba activate r_env mamba install -c conda-forge -c bioconda \ bioconductor-marray \ bioconductor-rgraphviz \ r-plyr r-gplots r-reshape2 r-ggplot2 r-scales r-openxlsx r-rcurl r-xml \ -y mamba install -c conda-forge -c bioconda \ r-plyr r-gplots r-reshape2 r-ggplot2 r-scales r-openxlsx \ bioconductor-marray bioconductor-rgraphviz \ -y #mkdir summaries heatmap_all_WaGa+4_MKL-1 mkdir results_WaGa_EXCLUDED results_MKL-1 summaries_WaGa summaries_MKL-1 heatmap_WaGa heatmap_MKL-1 #! EXCLUDE some isolates since they have total different pattern or due to bad quality --> outliner, until now only one sample, namely nf657 from WaGa wt EV: sudo mv results/nf657* results_WaGa_EXCLUDED/ sudo mv results/nf780* results_MKL-1/ sudo mv results/nf796* results_MKL-1/ sudo mv results/nf797* results_MKL-1/ sudo mv results/nf655* results_MKL-1/ for sample in 2404_MKL1_wt_EVs 2608_MKL1_wt_EVs 2608_MKL1_sT_DMSO 2701_MKL1_sT_DMSO 2802_MKL1_sT_DMSO 2608_MKL1_sT_Dox 2701_MKL1_sT_Dox 2802_MKL1_sT_Dox 2608_MKL1_scr_DMSO 2701_MKL1_scr_DMSO 2802_MKL1_scr_DMSO 2608_MKL1_scr_Dox 2701_MKL1_scr_Dox 2802_MKL1_scr_Dox; do echo "sudo mv results/${sample}* results_MKL-1/" done #Following our initial QC, I noticed that one of the MKL-1 wt-EV samples (nf655) is a clear outlier, clustering far apart from the other two wt-EV replicates in the PCoA plots. I recommend removing nf655 from the downstream MKL-1 analysis, which is similar to our earlier analysis for MKL-1, in which we removed the outlier nf657. Please see the attached figures for reference. mv results_MKL-1/nf655* results_MKL-1_EXCLUDED/ (r_env) jhuang@WS-2290C:~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/exceRpt-master$ R #WARNING: need to reload the R-script after each change of the script. source("mergePipelineRuns_functions.R") processSamplesInDir("../results_WaGa/", "../summaries_WaGa") processSamplesInDir("../results_MKL-1/", "../summaries_MKL-1") #mkdir heatmap_WaGa; cp summaries_WaGa/*.RData heatmap_WaGa; rm heatmap_WaGa/exceRpt_sampleGroupDefinitions.txt; source("mergePipelineRuns_functions_addSampleGroupInfo_WaGa.R") processSamplesInDir("../results_WaGa/", "../heatmap_WaGa") #mkdir heatmap_MKL-1; cp summaries_MKL-1/*.RData heatmap_MKL-1; rm heatmap_MKL-1/exceRpt_sampleGroupDefinitions.txt; source("mergePipelineRuns_functions_addSampleGroupInfo_MKL-1.R") processSamplesInDir("../results_MKL-1/", "../heatmap_MKL-1") #!!!!! IMPORTANT: REPORT heatmap_MKL-1/exceRpt_DiagnosticPlots.pdf and heatmap_MKL-1/mapping_heatmap3.pdf (They are almost the same, mapping_heatmap3.pdf is better due to bigger font size) !!!! #CONSIDERING_TO_DEL_nf774 since it is very far to another two samples (MAYBE BETTER NOT DO THIS, SINCE I HAVE TO GENERATE PCA- and MANHATTAN PLOTS!!): now the sample nf774 was kept in the WaGa results. #~/Tools/csv2xls-0.4/csv_to_xls.py exceRpt_miRNA_ReadsPerMillion.txt exceRpt_tRNA_ReadsPerMillion.txt exceRpt_piRNA_ReadsPerMillion.txt -d$'\t' -o exceRpt_results_detailed.xls # Report summaries_WaGa/exceRpt_mapping_heatmaps_WaGa.xlsx or summaries_MKL-1/exceRpt_mapping_heatmaps_MKL-1.xlsx; # summaries_WaGa/exceRpt_results_detailed_WaGa.xls or summaries_MKL-1/exceRpt_results_detailed_MKL-1.xls; # heatmap_WaGa/mapping_heatmap3_WaGa.pdf or heatmap_MKL-1/mapping_heatmap3_MKL-1.pdf -
Downstream analyis using R for miRNAs (17 WaGa samples)
#Input file #exceRpt_miRNA_ReadCounts.txt #exceRpt_piRNA_ReadCounts.txt ## WaGa experimental groups (scr = scramble control; sT = target knockdown) #WaGa_scr_DMSO_EV (nf933, nf938, nf973) #WaGa_scr_Dox_EV (nf934, nf939, nf974) #WaGa_sT_DMSO_EV (nf931, nf936, nf971) #WaGa_sT_Dox_EV (nf932, nf937, nf972) # ## WaGa wild-type controls #WaGa_wt_cells (nf774, nf961, nf962) #WaGa_wt_EV (nf930, nf935) cd ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/summaries_WaGa mamba activate r_env R #BiocManager::install("AnnotationDbi") #BiocManager::install("clusterProfiler") #BiocManager::install(c("ReactomePA","org.Hs.eg.db")) #BiocManager::install("limma") #BiocManager::install("sva") #install.packages("writexl") #install.packages("openxlsx") library("AnnotationDbi") library("clusterProfiler") library("ReactomePA") library("org.Hs.eg.db") library(DESeq2) library(gplots) library(limma) library(sva) #library(writexl) #d.raw_with_rownames <- cbind(RowNames = rownames(d.raw), d.raw); write_xlsx(d.raw, path = "d_raw.xlsx"); library(openxlsx) d.raw<- read.delim2("exceRpt_miRNA_ReadCounts.txt",sep="\t", header=TRUE, row.names=1) # Desired column order desired_order <- c( "nf933", "nf938", "nf973", "nf934", "nf939", "nf974", "nf931", "nf936", "nf971", "nf932", "nf937", "nf972", "nf774", "nf961", "nf962", "nf930", "nf935" ) # Reorder columns d.raw <- d.raw[, desired_order] setdiff(desired_order, colnames(d.raw)) # Shows missing or misnamed columns #sapply(d.raw, is.numeric) d.raw[] <- lapply(d.raw, as.numeric) #d.raw[] <- lapply(d.raw, function(x) as.numeric(as.character(x))) d.raw <- round(d.raw) write.csv(d.raw, file ="d_raw.csv") write.xlsx(d.raw, file = "d_raw.xlsx", rowNames = TRUE) # ------ Code sent to Ute ------ #d.raw <- read.delim2("d_raw.csv",sep=",", header=TRUE, row.names=1) Cell_or_EV = as.factor(c("EV","EV","EV", "EV","EV","EV", "EV","EV","EV", "EV","EV","EV", "Cell","Cell","Cell", "EV","EV")) replicates = as.factor(c("WaGa_scr_DMSO_EV","WaGa_scr_DMSO_EV","WaGa_scr_DMSO_EV", "WaGa_scr_Dox_EV","WaGa_scr_Dox_EV","WaGa_scr_Dox_EV", "WaGa_sT_DMSO_EV","WaGa_sT_DMSO_EV","WaGa_sT_DMSO_EV", "WaGa_sT_Dox_EV","WaGa_sT_Dox_EV","WaGa_sT_Dox_EV", "WaGa_wt_cells", "WaGa_wt_cells","WaGa_wt_cells", "WaGa_wt_EV", "WaGa_wt_EV")) ids = as.factor(c( "nf933", "nf938", "nf973", "nf934", "nf939", "nf974", "nf931", "nf936", "nf971", "nf932", "nf937", "nf972", "nf774", "nf961", "nf962", "nf930", "nf935")) cData = data.frame(row.names=colnames(d.raw), replicates=replicates, ids=ids, Cell_or_EV=Cell_or_EV) dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) # Filter low-count miRNAs dds <- dds[ rowSums(counts(dds)) > 10, ] rld <- rlogTransformation(dds) # -- before pca -- png("pca.png", 1200, 800) plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) #plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() png("pca2.png", 1200, 800) #plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() # Batch Effect Removal Methods (Non-batch effect removal applied!) #### STEP2: DEGs #### #- Heatmap untreated/wt vs parental; 1x for WaGa cell line #- Volcano plot untreated/wt vs parental; 1x for WaGa cell line #- Manhattan plot miRNAs; 1x for WaGa cell line #- Distribution of different small RNA species untreated/wt and parental; 1x for WaGa cell line #- Motif analysis: identify RNA-binding proteins that may regulate small RNA loading; 1x for WaGa cell line #convert bam to bigwig using deepTools by feeding inverse of DESeq’s size Factor sizeFactors(dds) #NULL dds <- estimateSizeFactors(dds) sizeFactors(dds) normalized_counts <- counts(dds, normalized=TRUE) write.table(normalized_counts, file="normalized_counts.txt", sep="\t", quote=F, col.names=NA) write.xlsx(normalized_counts, file = "normalized_counts.xlsx", rowNames = TRUE) dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) dds$replicates <- relevel(dds$replicates, "WaGa_wt_cells") dds = DESeq(dds, betaPrior=FALSE) #default betaPrior is FALSE resultsNames(dds) clist <- c("WaGa_wt_EV_vs_WaGa_wt_cells") #NOTE that the results sent to Ute is |padj|<=0.1. for (i in clist) { contrast = paste("replicates", i, sep="_") res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] #https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#why-are-some-p-values-set-to-na res$padj <- ifelse(is.na(res$padj), 1, res$padj) res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(i, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(i, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(i, "down.txt", sep="-")) } ~/Tools/csv2xls-0.4/csv_to_xls.py \ WaGa_wt_EV_vs_WaGa_wt_cells-all.txt \ WaGa_wt_EV_vs_WaGa_wt_cells-up.txt \ WaGa_wt_EV_vs_WaGa_wt_cells-down.txt \ -d$',' -o WaGa_wt_EV_vs_WaGa_wt_cells.xls; # ------------------- volcano_plot ------------------- library(ggplot2) library(ggrepel) geness_res <- read.csv(file = paste("WaGa_wt_EV_vs_WaGa_wt_cells", "all.txt", sep="-"), row.names=1) external_gene_name <- rownames(geness_res) geness_res <- cbind(geness_res, external_gene_name) #top_g are from ids top_g <- c("hsa-let-7b-5p","hsa-let-7g-5p","hsa-let-7i-5p","hsa-miR-103a-3p","hsa-miR-107","hsa-miR-1224-5p","hsa-miR-122-5p","hsa-miR-1226-5p","hsa-miR-1246","hsa-miR-127-3p","hsa-miR-1290","hsa-miR-130a-3p","hsa-miR-139-3p","hsa-miR-141-3p","hsa-miR-143-3p","hsa-miR-148b-3p","hsa-miR-155-5p","hsa-miR-15a-5p","hsa-miR-17-5p","hsa-miR-184","hsa-miR-18a-3p","hsa-miR-18a-5p","hsa-miR-190a-5p","hsa-miR-191-5p","hsa-miR-193b-5p","hsa-miR-197-5p","hsa-miR-200a-3p","hsa-miR-200b-5p","hsa-miR-206","hsa-miR-20a-5p","hsa-miR-210-3p","hsa-miR-2110","hsa-miR-21-5p","hsa-miR-218-5p","hsa-miR-219a-1-3p","hsa-miR-221-3p","hsa-miR-23b-3p","hsa-miR-27a-3p","hsa-miR-27b-3p","hsa-miR-27b-5p","hsa-miR-28-3p","hsa-miR-30a-5p","hsa-miR-30c-5p","hsa-miR-30e-5p","hsa-miR-3127-5p","hsa-miR-3131","hsa-miR-3180|hsa-miR-3180-3p","hsa-miR-320a","hsa-miR-320b","hsa-miR-320c","hsa-miR-320d","hsa-miR-330-3p","hsa-miR-335-3p","hsa-miR-33b-5p","hsa-miR-340-5p","hsa-miR-342-5p","hsa-miR-3605-5p","hsa-miR-361-3p","hsa-miR-365a-5p","hsa-miR-374b-5p","hsa-miR-378i","hsa-miR-379-5p","hsa-miR-3940-5p","hsa-miR-409-3p","hsa-miR-411-5p","hsa-miR-423-3p","hsa-miR-423-5p","hsa-miR-4286","hsa-miR-429","hsa-miR-432-5p","hsa-miR-4326","hsa-miR-451a","hsa-miR-4520-3p","hsa-miR-454-3p","hsa-miR-4646-5p","hsa-miR-4667-5p","hsa-miR-4748","hsa-miR-483-5p","hsa-miR-486-5p","hsa-miR-5010-5p","hsa-miR-504-3p","hsa-miR-5187-5p","hsa-miR-590-3p","hsa-miR-6128","hsa-miR-625-5p","hsa-miR-6726-5p","hsa-miR-6730-5p","hsa-miR-676-3p","hsa-miR-6767-5p","hsa-miR-6777-5p","hsa-miR-6780a-5p","hsa-miR-6794-5p","hsa-miR-6817-3p","hsa-miR-708-5p","hsa-miR-7-5p","hsa-miR-766-5p","hsa-miR-7854-3p","hsa-miR-873-3p","hsa-miR-885-3p","hsa-miR-92b-5p","hsa-miR-93-5p","hsa-miR-937-3p","hsa-miR-9-5p","hsa-miR-98-5p") subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)) geness_res$Color <- "NS or log2FC < 2.0" geness_res$Color[geness_res$pvalue < 0.05] <- "P < 0.05" geness_res$Color[geness_res$padj < 0.05] <- "P-adj < 0.05" geness_res$Color[abs(geness_res$log2FoldChange) < 2.0] <- "NS or log2FC < 2.0" write.csv(geness_res, "WaGa_wt_EV_vs_WaGa_wt_cells_with_Category.csv") geness_res$invert_P <- (-log10(geness_res$pvalue)) * sign(geness_res$log2FoldChange) geness_res <- geness_res[, -1*ncol(geness_res)] png("WaGa_wt_EV_vs_WaGa_wt_cells.png",width=1200, height=1400) #svg("WaGa_wt_EV_vs_WaGa_wt_cells.svg",width=12, height=14) ggplot(geness_res, aes(x = log2FoldChange, y = -log10(pvalue), color = Color, label = external_gene_name)) + geom_vline(xintercept = c(2.0, -2.0), lty = "dashed") + geom_hline(yintercept = -log10(0.05), lty = "dashed") + geom_point() + labs(x = "log2(FC)", y = "Significance, -log10(P)", color = "Significance") + scale_color_manual(values = c("P < 0.05"="orange","P-adj < 0.05"="red","NS or log2FC < 2.0"="darkgray"),guide = guide_legend(override.aes = list(size = 4))) + scale_y_continuous(expand = expansion(mult = c(0,0.05))) + geom_text_repel(data = subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)), size = 4, point.padding = 0.15, color = "black", min.segment.length = .1, box.padding = .2, lwd = 2) + theme_bw(base_size = 16) + theme(legend.position = "bottom") dev.off() # ---------------------------------------- # ----------- manhattan_plot ------------- Rscript manhattan_plot_Carmen_custom_labels.R #exceRpt_miRNA_ReadCounts.txt -
Downstream analyis using R for miRNAs (17 MKL-1 samples)
#Input file #exceRpt_miRNA_ReadCounts.txt #exceRpt_piRNA_ReadCounts.txt #MKL-1_sT_DMSO_EV ("X2608_MKL1_sT_DMSO","X2701_MKL1_sT_DMSO","X2802_MKL1_sT_DMSO") #MKL-1_sT_Dox_EV ("X2608_MKL1_sT_Dox","X2701_MKL1_sT_Dox","X2802_MKL1_sT_Dox") #MKL-1_scr_DMSO_EV ("X2608_MKL1_scr_DMSO","X2701_MKL1_scr_DMSO","X2802_MKL1_scr_DMSO") #MKL-1_scr_Dox_EV ()"X2608_MKL1_scr_Dox","X2701_MKL1_scr_Dox","X2802_MKL1_scr_Dox") #MKL-1_wt_cells ("nf780","nf796","nf797") #MKL-1_wt_EV ("X2404_MKL1_wt_EVs","X2608_MKL1_wt_EVs") cd ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/summaries_MKL-1 mamba activate r_env R #BiocManager::install("AnnotationDbi") #BiocManager::install("clusterProfiler") #BiocManager::install(c("ReactomePA","org.Hs.eg.db")) #BiocManager::install("limma") #BiocManager::install("sva") #install.packages("writexl") #install.packages("openxlsx") library("AnnotationDbi") library("clusterProfiler") library("ReactomePA") library("org.Hs.eg.db") library(DESeq2) library(gplots) library(limma) library(sva) #library(writexl) #d.raw_with_rownames <- cbind(RowNames = rownames(d.raw), d.raw); write_xlsx(d.raw, path = "d_raw.xlsx"); library(openxlsx) d.raw<- read.delim2("exceRpt_miRNA_ReadCounts.txt",sep="\t", header=TRUE, row.names=1) # Desired column order desired_order <- c( "X2608_MKL1_sT_DMSO","X2701_MKL1_sT_DMSO","X2802_MKL1_sT_DMSO", "X2608_MKL1_sT_Dox","X2701_MKL1_sT_Dox","X2802_MKL1_sT_Dox", "X2608_MKL1_scr_DMSO","X2701_MKL1_scr_DMSO","X2802_MKL1_scr_DMSO", "X2608_MKL1_scr_Dox","X2701_MKL1_scr_Dox","X2802_MKL1_scr_Dox", "nf780","nf796","nf797", "X2404_MKL1_wt_EVs","X2608_MKL1_wt_EVs" ) # Reorder columns d.raw <- d.raw[, desired_order] setdiff(desired_order, colnames(d.raw)) # Shows missing or misnamed columns #sapply(d.raw, is.numeric) d.raw[] <- lapply(d.raw, as.numeric) #d.raw[] <- lapply(d.raw, function(x) as.numeric(as.character(x))) d.raw <- round(d.raw) write.csv(d.raw, file ="d_raw.csv") write.xlsx(d.raw, file = "d_raw.xlsx", rowNames = TRUE) #d.raw <- read.delim2("d_raw.csv",sep=",", header=TRUE, row.names=1) Cell_or_EV = as.factor(c("EV","EV","EV", "EV","EV","EV", "EV","EV","EV", "EV","EV","EV", "Cell","Cell","Cell", "EV","EV")) replicates = as.factor(c("MKL-1_sT_DMSO_EV","MKL-1_sT_DMSO_EV","MKL-1_sT_DMSO_EV", "MKL-1_sT_Dox_EV","MKL-1_sT_Dox_EV","MKL-1_sT_Dox_EV", "MKL-1_scr_DMSO_EV","MKL-1_scr_DMSO_EV","MKL-1_scr_DMSO_EV", "MKL-1_scr_Dox_EV","MKL-1_scr_Dox_EV","MKL-1_scr_Dox_EV", "MKL-1_wt_cells", "MKL-1_wt_cells","MKL-1_wt_cells", "MKL-1_wt_EV","MKL-1_wt_EV")) ids = as.factor(c("X2608_MKL1_sT_DMSO","X2701_MKL1_sT_DMSO","X2802_MKL1_sT_DMSO", "X2608_MKL1_sT_Dox","X2701_MKL1_sT_Dox","X2802_MKL1_sT_Dox", "X2608_MKL1_scr_DMSO","X2701_MKL1_scr_DMSO","X2802_MKL1_scr_DMSO", "X2608_MKL1_scr_Dox","X2701_MKL1_scr_Dox","X2802_MKL1_scr_Dox", "nf780","nf796","nf797", "X2404_MKL1_wt_EVs","X2608_MKL1_wt_EVs")) cData = data.frame(row.names=colnames(d.raw), replicates=replicates, ids=ids, Cell_or_EV=Cell_or_EV) dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) # Filter low-count miRNAs dds <- dds[ rowSums(counts(dds)) > 10, ] rld <- rlogTransformation(dds) # -- before pca -- png("pca.png", 1200, 800) plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) #plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() png("pca2.png", 1200, 800) #plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() # Batch Effect Removal Methods (Non-batch effect removal applied!) #### STEP2: DEGs #### #- Heatmap untreated/wt vs parental; 1x for WaGa cell line #- Volcano plot untreated/wt vs parental; 1x for WaGa cell line #- Manhattan plot miRNAs; 1x for WaGa cell line #- Distribution of different small RNA species untreated/wt and parental; 1x for WaGa cell line #- Motif analysis: identify RNA-binding proteins that may regulate small RNA loading; 1x for WaGa cell line #convert bam to bigwig using deepTools by feeding inverse of DESeq’s size Factor sizeFactors(dds) #NULL dds <- estimateSizeFactors(dds) sizeFactors(dds) normalized_counts <- counts(dds, normalized=TRUE) write.table(normalized_counts, file="normalized_counts.txt", sep="\t", quote=F, col.names=NA) write.xlsx(normalized_counts, file = "normalized_counts.xlsx", rowNames = TRUE) dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) dds$replicates <- relevel(dds$replicates, "MKL-1_wt_cells") dds = DESeq(dds, betaPrior=FALSE) #default betaPrior is FALSE resultsNames(dds) clist <- c("MKL.1_wt_EV_vs_MKL.1_wt_cells") #NOTE that the results sent to Ute is |padj|<=0.1. for (i in clist) { contrast = paste("replicates", i, sep="_") res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] #https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#why-are-some-p-values-set-to-na res$padj <- ifelse(is.na(res$padj), 1, res$padj) res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(i, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(i, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(i, "down.txt", sep="-")) } ~/Tools/csv2xls-0.4/csv_to_xls.py \ MKL.1_wt_EV_vs_MKL.1_wt_cells-all.txt \ MKL.1_wt_EV_vs_MKL.1_wt_cells-up.txt \ MKL.1_wt_EV_vs_MKL.1_wt_cells-down.txt \ -d$',' -o MKL.1_wt_EV_vs_MKL.1_wt_cells.xls; # ------------------- volcano_plot ------------------- library(ggplot2) library(ggrepel) geness_res <- read.csv(file = paste("MKL.1_wt_EV_vs_MKL.1_wt_cells", "all.txt", sep="-"), row.names=1) external_gene_name <- rownames(geness_res) geness_res <- cbind(geness_res, external_gene_name) #top_g are from ids top_g <- c("hsa-miR-203a-3p","hsa-miR-6850-5p","hsa-miR-4511","hsa-miR-5187-5p","hsa-miR-133b","hsa-miR-1246","hsa-miR-625-3p","hsa-miR-6741-3p","hsa-miR-192-5p","hsa-miR-10b-5p","hsa-miR-885-5p","hsa-miR-30e-3p","hsa-miR-101-3p","hsa-miR-1307-5p","hsa-miR-95-3p","hsa-miR-889-3p","hsa-miR-206","hsa-miR-301a-3p","hsa-miR-1-3p","hsa-let-7c-5p","hsa-miR-196a-5p","hsa-let-7f-5p","hsa-let-7e-5p","hsa-miR-30c-5p","hsa-miR-30a-3p","hsa-miR-146b-5p","hsa-miR-25-3p","hsa-miR-182-5p","hsa-miR-98-5p","hsa-let-7a-5p","hsa-miR-149-5p","hsa-miR-148a-3p","hsa-miR-873-3p","hsa-miR-19b-3p","hsa-miR-320c","hsa-miR-375","hsa-miR-30a-5p","hsa-miR-877-5p","hsa-miR-34a-5p","hsa-miR-324-5p","hsa-miR-652-3p","hsa-miR-342-5p","hsa-miR-7706","hsa-miR-361-3p","hsa-miR-361-5p","hsa-miR-1180-3p","hsa-miR-217","hsa-miR-1307-3p","hsa-miR-1908-5p","hsa-miR-15b-5p","hsa-miR-92b-5p","hsa-miR-484","hsa-miR-197-3p","hsa-miR-200c-3p","hsa-miR-671-5p","hsa-miR-339-5p","hsa-miR-1301-3p","hsa-miR-769-5p","hsa-miR-328-3p","hsa-miR-93-5p","hsa-miR-103a-3p") subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)) geness_res$Color <- "NS or log2FC < 2.0" geness_res$Color[geness_res$pvalue < 0.05] <- "P < 0.05" geness_res$Color[geness_res$padj < 0.05] <- "P-adj < 0.05" geness_res$Color[abs(geness_res$log2FoldChange) < 2.0] <- "NS or log2FC < 2.0" write.csv(geness_res, "MKL.1_wt_EV_vs_MKL.1_wt_cells_with_Category.csv") geness_res$invert_P <- (-log10(geness_res$pvalue)) * sign(geness_res$log2FoldChange) geness_res <- geness_res[, -1*ncol(geness_res)] png("MKL.1_wt_EV_vs_MKL.1_wt_cells.png",width=1200, height=1400) #svg("MKL.1_wt_EV_vs_MKL.1_wt_cells.svg",width=12, height=14) ggplot(geness_res, aes(x = log2FoldChange, y = -log10(pvalue), color = Color, label = external_gene_name)) + geom_vline(xintercept = c(2.0, -2.0), lty = "dashed") + geom_hline(yintercept = -log10(0.05), lty = "dashed") + geom_point() + labs(x = "log2(FC)", y = "Significance, -log10(P)", color = "Significance") + scale_color_manual(values = c("P < 0.05"="orange","P-adj < 0.05"="red","NS or log2FC < 2.0"="darkgray"),guide = guide_legend(override.aes = list(size = 4))) + scale_y_continuous(expand = expansion(mult = c(0,0.05))) + geom_text_repel(data = subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)), size = 4, point.padding = 0.15, color = "black", min.segment.length = .1, box.padding = .2, lwd = 2) + theme_bw(base_size = 16) + theme(legend.position = "bottom") dev.off() # ---------------------------------------- # ----------- manhattan_plot ------------- Rscript manhattan_plot_Carmen_custom_labels.R #exceRpt_miRNA_ReadCounts.txt
Until now, done and sent!
- Raw count data (d_raw_MKL-1.xlsx): Contains the raw, unnormalized read counts for all miRNAs.
- Mapping heatmap (mapping_heatmap3_MKL-1.pdf)
- Volcano plot (MKL.1_wt_EV_vs_MKL.1_wt_cells.png and .svg)
- PCA plot (pca_MKL-1.png)
- Manhattan plot and data (manhattan_plot_MKL1_vs_EV.png, .svg, and manhattan_plot_MKL1_data.xlsx)
-
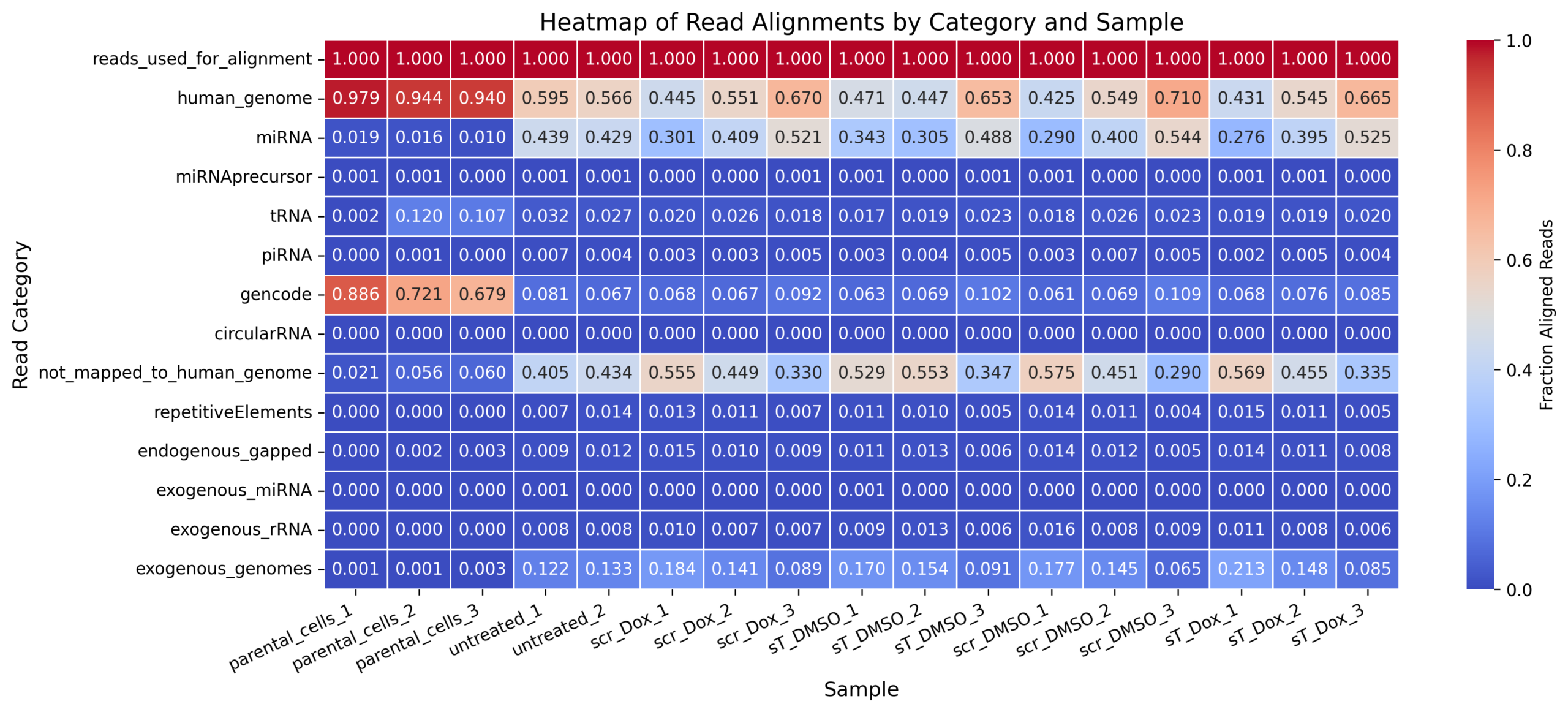
Draw distribution_heatmap.png for MKL-1 samples sent afterwards
# -- R-code -- # Load required library library(dplyr) library(openxlsx) # The numbers are extracted manually from summaries_MKL-1/mapping_heatmap3.pdf with the samples ordered by # sampleID sampleGroup sampleGroup2 # nf780 MKL-1 wt cells "parental_cells_1" # nf796 MKL-1 wt cells "parental_cells_2" # nf797 MKL-1 wt cells "parental_cells_3" # 2608_MKL1_sT_Dox MKL-1 sT Dox EV "sT_Dox_1" # 2701_MKL1_scr_DMSO MKL-1 scr DMSO EV "scr_DMSO_1" # 2701_MKL1_sT_DMSO MKL-1 sT DMSO EV "sT_DMSO_1" # 2608_MKL1_sT_DMSO MKL-1 sT DMSO EV "sT_DMSO_2" # 2701_MKL1_scr_Dox MKL-1 scr Dox EV "scr_Dox_1" # 2404_MKL1_wt_EVs MKL-1 wt EV "untreated_1" # 2608_MKL1_wt_EVs MKL-1 wt EV "untreated_2" # 2701_MKL1_sT_Dox MKL-1 sT Dox EV "sT_Dox_2" # 2608_MKL1_scr_DMSO MKL-1 scr DMSO EV "scr_DMSO_2" # 2608_MKL1_scr_Dox MKL-1 scr Dox EV "scr_Dox_2" # 2802_MKL1_scr_DMSO MKL-1 scr DMSO EV "scr_DMSO_3" # 2802_MKL1_sT_DMSO MKL-1 sT DMSO EV "sT_DMSO_3" # 2802_MKL1_scr_Dox MKL-1 scr Dox EV "scr_Dox_3" # 2802_MKL1_sT_Dox MKL-1 sT Dox EV "sT_Dox_3" # Original data matrix (Note that the following is the complete table including tRNA_sense and tRNA_antisense ..., the code summing the sense and antisense numbers and resulting in total numbers) data_orig <- matrix(c(100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 100.0, 97.9, 94.4, 94.0, 43.1, 42.5, 47.1, 44.7, 44.5, 59.5, 56.6, 54.5, 54.9, 55.1, 71.0, 65.3, 67.0, 66.5, 1.9, 1.6, 1.0, 27.6, 29.0, 34.3, 30.5, 30.1, 43.9, 42.9, 39.5, 40.0, 40.9, 54.4, 48.8, 52.1, 52.5, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.1, 0.1, 0.0, 0.1, 0.1, 0.1, 0.0, 0.0, 0.1, 0.1, 0.1, 0.0, 0.0, 0.0, 0.1, 0.1, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.2, 12.0, 10.7, 1.9, 1.8, 1.7, 1.9, 2.0, 3.2, 2.7, 1.9, 2.6, 2.6, 2.3, 2.3, 1.8, 2.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.1, 0.0, 0.2, 0.3, 0.3, 0.4, 0.3, 0.7, 0.4, 0.5, 0.7, 0.3, 0.5, 0.5, 0.5, 0.4, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 88.5, 71.9, 67.5, 6.6, 5.9, 6.0, 6.6, 6.5, 7.7, 6.4, 7.4, 6.6, 6.5, 10.7, 10.0, 9.0, 8.3, 0.1, 0.2, 0.4, 0.2, 0.2, 0.3, 0.3, 0.3, 0.4, 0.3, 0.2, 0.3, 0.2, 0.2, 0.2, 0.2, 0.2, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 2.1, 5.6, 6.0, 56.9, 57.5, 52.9, 55.3, 55.5, 40.5, 43.4, 45.5, 45.1, 44.9, 29.0, 34.7, 33.0, 33.5, 0.0, 0.0, 0.0, 1.5, 1.4, 1.1, 1.0, 1.3, 0.7, 1.4, 1.1, 1.1, 1.1, 0.4, 0.5, 0.7, 0.5, 0.0, 0.2, 0.3, 1.4, 1.4, 1.1, 1.3, 1.5, 0.9, 1.2, 1.1, 1.2, 1.0, 0.5, 0.6, 0.9, 0.8, 0.0, 0.0, 0.0, 0.0, 0.0, 0.1, 0.0, 0.0, 0.1, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 0.0, 1.1, 1.6, 0.9, 1.3, 1.0, 0.8, 0.8, 0.8, 0.8, 0.7, 0.9, 0.6, 0.7, 0.6, 0.1, 0.1, 0.3, 21.3, 17.7, 17.0, 15.4, 18.4, 12.2, 13.3, 14.8, 14.5, 14.1, 6.5, 9.1, 8.9, 8.5), nrow = 20, byrow = TRUE) # Original vectors samples_orig <- c("parental_cells_1", "parental_cells_2", "parental_cells_3", "sT_Dox_1", "scr_DMSO_1", "sT_DMSO_1", "sT_DMSO_2", "scr_Dox_1", "untreated_1", "untreated_2", "sT_Dox_2", "scr_DMSO_2", "scr_Dox_2", "scr_DMSO_3", "sT_DMSO_3", "scr_Dox_3", "sT_Dox_3") categories_orig <- c("reads_used_for_alignment", "genome", "miRNA_sense", "miRNA_antisense", "miRNAprecursor_sense", "miRNAprecursor_antisense", "tRNA_sense", "tRNA_antisense", "piRNA_sense", "piRNA_antisense", "gencode_sense", "gencode_antisense", "circularRNA_sense", "circularRNA_antisense", "not_mapped_to_genome_or_libs", "repetitiveElements", "endogenous_gapped", "exogenous_miRNA", "exogenous_rRNA", "exogenous_genomes") # Provided samples and categories (desired order and format) samples <- c("parental_cells_1","parental_cells_2","parental_cells_3", "untreated_1","untreated_2", "scr_Dox_1","scr_Dox_2","scr_Dox_3", "sT_DMSO_1","sT_DMSO_2","sT_DMSO_3", "scr_DMSO_1","scr_DMSO_2","scr_DMSO_3", "sT_Dox_1","sT_Dox_2","sT_Dox_3") categories <- c("reads_used_for_alignment", "genome", "miRNA", "miRNAprecursor", "tRNA", "piRNA", "gencode", "circularRNA", "not_mapped_to_genome_or_libs", "repetitiveElements", "endogenous_gapped", "exogenous_miRNA", "exogenous_rRNA", "exogenous_genomes") rownames(data_orig) <- categories_orig colnames(data_orig) <- samples_orig # Collapse sense/antisense merge_rows <- function(prefix) { row1 <- paste0(prefix, "_sense") row2 <- paste0(prefix, "_antisense") if (row1 %in% rownames(data_orig) && row2 %in% rownames(data_orig)) { return(data_orig[row1, ] + data_orig[row2, ]) } else if (row1 %in% rownames(data_orig)) { return(data_orig[row1, ]) } else { return(rep(0, ncol(data_orig))) } } # Construct merged data data_merged <- rbind( reads_used_for_alignment = data_orig["reads_used_for_alignment", ], genome = data_orig["genome", ], miRNA = merge_rows("miRNA"), miRNAprecursor = merge_rows("miRNAprecursor"), tRNA = merge_rows("tRNA"), piRNA = merge_rows("piRNA"), gencode = merge_rows("gencode"), circularRNA = merge_rows("circularRNA"), not_mapped_to_genome_or_libs = data_orig["not_mapped_to_genome_or_libs", ], repetitiveElements = data_orig["repetitiveElements", ], endogenous_gapped = data_orig["endogenous_gapped", ], exogenous_miRNA = data_orig["exogenous_miRNA", ], exogenous_rRNA = data_orig["exogenous_rRNA", ], exogenous_genomes = data_orig["exogenous_genomes", ] ) # Reorder columns to match desired sample order data_final <- data_merged[, samples[samples %in% colnames(data_merged)]] #genome --> human_genome, not_mapped_to_genome_or_libs --> not_mapped_to_human_genome rownames(data_final)[rownames(data_final) == "genome"] <- "human_genome" rownames(data_final)[rownames(data_final) == "not_mapped_to_genome_or_libs"] <- "not_mapped_to_human_genome" # Save to Excel write.xlsx(data_final, file = "distribution_heatmap.xlsx", rowNames = TRUE) # -- Python-code -- python plot_distribution_heatmap.py distribution_heatmap.xlsx distribution_heatmap.png import pandas as pd import numpy as np import seaborn as sns import matplotlib.pyplot as plt ## Load data from Excel file file_path = "distribution_heatmap.xlsx" # Read Excel file, assuming first column is index (row labels) df = pd.read_excel(file_path, index_col=0) # The data is already in percentage format, convert to decimals if needed # If you want fractions (0-1 range), divide by 100 data = df.values / 100.0 # Get categories (row names) and samples (column names) categories = df.index.tolist() samples = df.columns.tolist() # Create DataFrame with proper structure df_plot = pd.DataFrame(data, index=categories, columns=samples) # Plot heatmap plt.figure(figsize=(14, 6)) sns.heatmap(df_plot, annot=True, cmap="coolwarm", fmt=".3f", linewidths=0.5, cbar_kws={'label': 'Fraction Aligned Reads'}) # Improve layout plt.title("Heatmap of Read Alignments by Category and Sample", fontsize=14) plt.xlabel("Sample", fontsize=12) plt.ylabel("Read Category", fontsize=12) plt.xticks(rotation=25, ha="right", fontsize=10) plt.yticks(rotation=0, fontsize=10) plt.tight_layout() # Save as PNG plt.savefig("distribution_heatmap.png", dpi=300, bbox_inches="tight") # Save as SVG (Vector) - Recommended for publications/editing plt.savefig("distribution_heatmap.svg", bbox_inches="tight") # Show plot plt.show() -
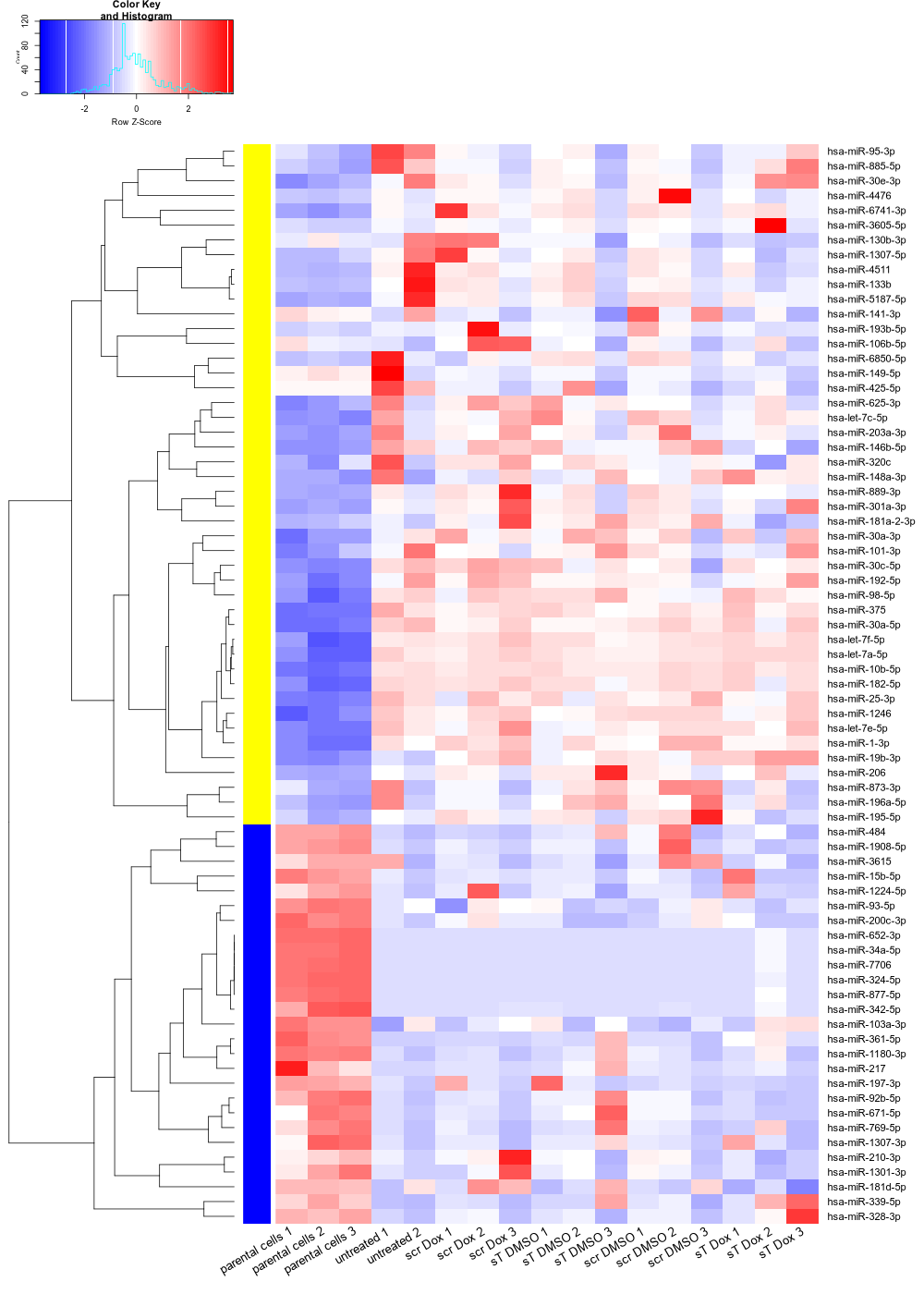
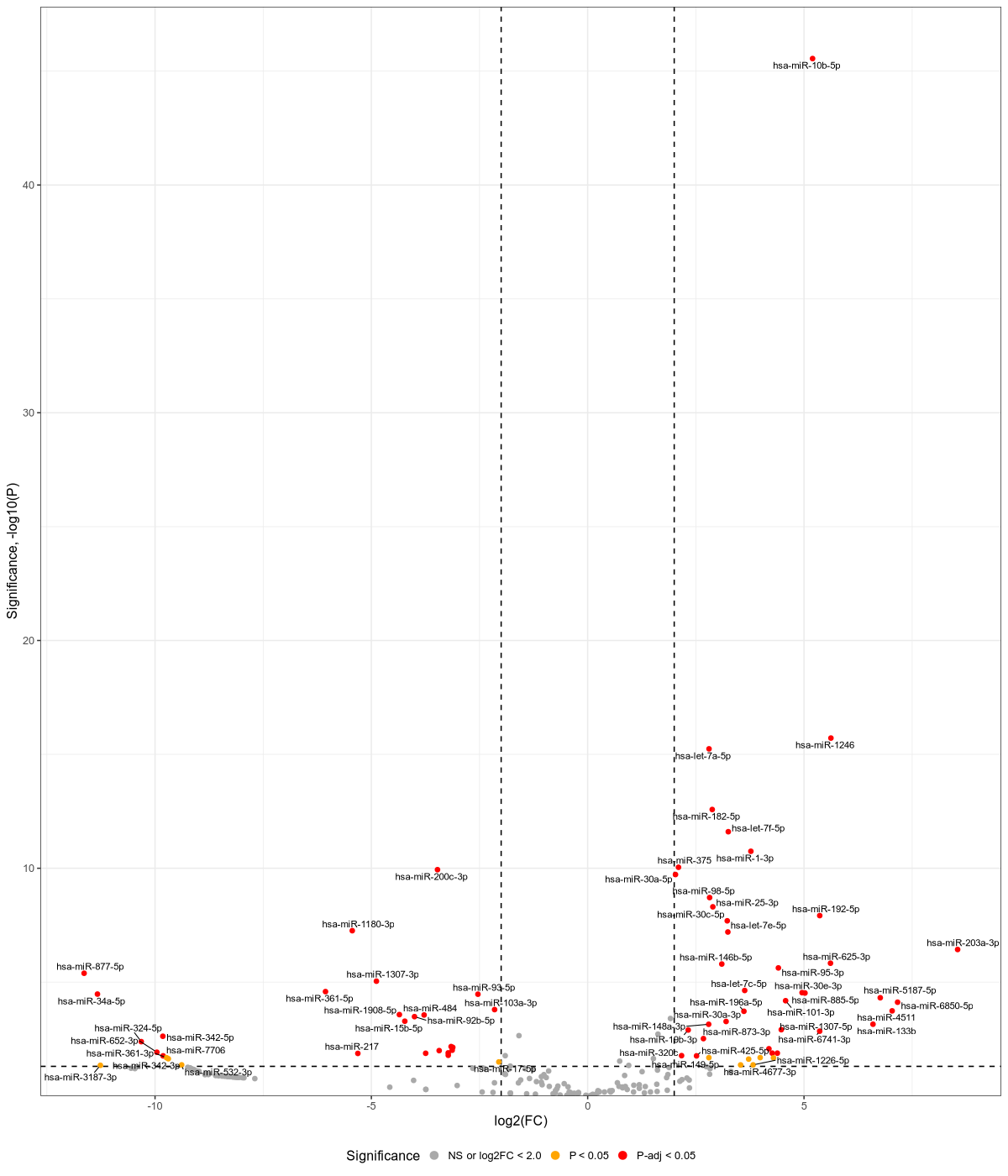
Draw differentially_expressed_miRNAs_heatmap.png sent afterwards (Namely downstream analyis using R for miRNAs)
#Input file #exceRpt_miRNA_ReadCounts.txt #exceRpt_piRNA_ReadCounts.txt #cd ~/DATA/Data_Ute_smallRNA_7/summaries_exo7 cd ~/DATA/Data_Ute_smallRNA_via_exceRpt_workspace/summaries_MKL-1 mamba activate r_env R #> .libPaths() #[1] "/home/jhuang/mambaforge/envs/r_env/lib/R/library" #BiocManager::install("AnnotationDbi") #BiocManager::install("clusterProfiler") #BiocManager::install(c("ReactomePA","org.Hs.eg.db")) #BiocManager::install("limma") #BiocManager::install("sva") #install.packages("writexl") #install.packages("openxlsx") library("AnnotationDbi") library("clusterProfiler") library("ReactomePA") library("org.Hs.eg.db") library(DESeq2) library(gplots) library(limma) library(sva) #library(writexl) #d.raw_with_rownames <- cbind(RowNames = rownames(d.raw), d.raw); write_xlsx(d.raw, path = "d_raw.xlsx"); library(openxlsx) # 1. Load data d.raw <- read.delim2("exceRpt_miRNA_ReadCounts.txt", sep="\t", header=TRUE, row.names=1) # 2. Define the mapping from Original Column Names to Standardized Names # Ensure these keys EXACTLY match what is in colnames(d.raw) sample_map <- c( "nf780" = "parental_cells_1", "nf796" = "parental_cells_2", "nf797" = "parental_cells_3", "X2404_MKL1_wt_EVs" = "untreated_1", "X2608_MKL1_wt_EVs" = "untreated_2", "X2701_MKL1_scr_DMSO" = "scr_DMSO_1", "X2608_MKL1_scr_DMSO" = "scr_DMSO_2", "X2802_MKL1_scr_DMSO" = "scr_DMSO_3", "X2701_MKL1_scr_Dox" = "scr_Dox_1", "X2608_MKL1_scr_Dox" = "scr_Dox_2", "X2802_MKL1_scr_Dox" = "scr_Dox_3", "X2701_MKL1_sT_DMSO" = "sT_DMSO_1", "X2608_MKL1_sT_DMSO" = "sT_DMSO_2", "X2802_MKL1_sT_DMSO" = "sT_DMSO_3", "X2701_MKL1_sT_Dox" = "sT_Dox_1", "X2608_MKL1_sT_Dox" = "sT_Dox_2", "X2802_MKL1_sT_Dox" = "sT_Dox_3" ) # 3. Safe Renaming Strategy # Create a vector of new names for ALL current columns new_col_names <- colnames(d.raw) # Identify which current columns are in our map matched_indices <- match(colnames(d.raw), names(sample_map)) # Replace names where a match was found # matched_indices will be NA if no match is found found_mask <- !is.na(matched_indices) new_col_names[found_mask] <- sample_map[colnames(d.raw)[found_mask]] # Assign the new names back to the dataframe colnames(d.raw) <- new_col_names # Optional: Check if any expected samples were missing entirely missing_samples <- setdiff(names(sample_map), colnames(d.raw)) # Check against OLD names? No, check against original input # Better check: original_cols <- colnames(read.delim2("exceRpt_miRNA_ReadCounts.txt", sep="\t", header=TRUE, nrows=1)) missing_in_data <- setdiff(names(sample_map), original_cols) if(length(missing_in_data) > 0) { warning(paste("WARNING: The following samples from your map were NOT found in the file:", paste(missing_in_data, collapse=", "))) } # 4. Define the desired final order desired_order <- c( "parental_cells_1", "parental_cells_2", "parental_cells_3", "untreated_1", "untreated_2", "scr_Dox_1", "scr_Dox_2", "scr_Dox_3", "sT_DMSO_1", "sT_DMSO_2", "sT_DMSO_3", "scr_DMSO_1", "scr_DMSO_2", "scr_DMSO_3", "sT_Dox_1", "sT_Dox_2", "sT_Dox_3" ) # 5. Check for missing columns in the final desired set missing_final <- setdiff(desired_order, colnames(d.raw)) if (length(missing_final) > 0) { stop(paste("ERROR: Missing required columns after renaming:", paste(missing_final, collapse=", "))) } # 6. Subset and Reorder d.raw <- d.raw[, desired_order] # 7. Ensure numeric type and round d.raw[] <- lapply(d.raw, function(x) as.numeric(as.character(x))) d.raw <- round(d.raw) # 8. Save outputs write.csv(d.raw, file = "d_raw.csv", row.names = TRUE) write.xlsx(d.raw, file = "d_raw.xlsx", rowNames = TRUE) print("Processing complete. Files saved.") #d.raw <- read.delim2("d_raw.csv",sep=",", header=TRUE, row.names=1) parental_or_EV = as.factor(c("parental","parental","parental", "EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV","EV")) #batch = as.factor(c("Aug22","March25","March25", "Sep23","Sep23", "Sep23","Sep23","March25", "Sep23","Sep23","March25", "Sep23","Sep23","March25", "Sep23","Sep23","March25")) replicates = as.factor(c("parental_cells", "parental_cells", "parental_cells", "untreated", "untreated", "scr_Dox", "scr_Dox", "scr_Dox", "sT_DMSO", "sT_DMSO", "sT_DMSO", "scr_DMSO", "scr_DMSO", "scr_DMSO", "sT_Dox", "sT_Dox", "sT_Dox")) ids = as.factor(c( "parental_cells_1", "parental_cells_2", "parental_cells_3", "untreated_1", "untreated_2", "scr_Dox_1", "scr_Dox_2", "scr_Dox_3", "sT_DMSO_1", "sT_DMSO_2", "sT_DMSO_3", "scr_DMSO_1", "scr_DMSO_2", "scr_DMSO_3", "sT_Dox_1", "sT_Dox_2", "sT_Dox_3" )) cData = data.frame(row.names=colnames(d.raw), replicates=replicates, ids=ids, parental_or_EV=parental_or_EV) #dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates+batch) dds<-DESeqDataSetFromMatrix(countData=d.raw, colData=cData, design=~replicates) # Filter low-count miRNAs dds <- dds[ rowSums(counts(dds)) > 10, ] #1322-->903 rld <- rlogTransformation(dds) # -- before pca -- png("pca.png", 1200, 800) plotPCA(rld, intgroup=c("replicates")) #plotPCA(rld, intgroup = c("replicates", "batch")) #plotPCA(rld, intgroup = c("replicates", "ids")) #plotPCA(rld, "batch") dev.off() # Batch Effect Removal Methods (Non-batch effect removal applied!) #### STEP2: DEGs #### #- Heatmap untreated/wt vs parental; 1x for WaGa cell line #- Volcano plot untreated/wt vs parental; 1x for WaGa cell line #- Manhattan plot miRNAs; 1x for WaGa cell line #- Distribution of different small RNA species untreated/wt and parental; 1x for WaGa cell line #- Motif analysis: identify RNA-binding proteins that may regulate small RNA loading; 1x for WaGa cell line #convert bam to bigwig using deepTools by feeding inverse of DESeq’s size Factor sizeFactors(dds) #NULL dds <- estimateSizeFactors(dds) sizeFactors(dds) normalized_counts <- counts(dds, normalized=TRUE) write.table(normalized_counts, file="normalized_counts.txt", sep="\t", quote=F, col.names=NA) write.xlsx(normalized_counts, file = "normalized_counts.xlsx", rowNames = TRUE) #---- untreated, scr_Dox, sT_DMSO, scr_DMSO, sT_Dox to parental_cells ---- dds$replicates <- relevel(dds$replicates, "parental_cells") dds = DESeq(dds, betaPrior=FALSE) #default betaPrior is FALSE resultsNames(dds) clist <- c("untreated_vs_parental_cells") dds$replicates <- relevel(dds$replicates, "untreated") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_DMSO_vs_untreated", "scr_Dox_vs_untreated", "scr_DMSO_vs_untreated", "sT_Dox_vs_untreated") dds$replicates <- relevel(dds$replicates, "sT_DMSO") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_Dox_vs_sT_DMSO") dds$replicates <- relevel(dds$replicates, "scr_Dox") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_Dox_vs_scr_Dox") dds$replicates <- relevel(dds$replicates, "scr_DMSO") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("sT_Dox_vs_scr_DMSO") #NOTE that the results sent to Ute is |padj|<=0.1. for (i in clist) { contrast = paste("replicates", i, sep="_") res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] #https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html#why-are-some-p-values-set-to-na res$padj <- ifelse(is.na(res$padj), 1, res$padj) res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(i, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(i, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(i, "down.txt", sep="-")) } ~/Tools/csv2xls-0.4/csv_to_xls.py \ untreated_vs_parental_cells-all.txt \ untreated_vs_parental_cells-up.txt \ untreated_vs_parental_cells-down.txt \ -d$',' -o untreated_vs_parental_cells.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_DMSO_vs_untreated-all.txt \ sT_DMSO_vs_untreated-up.txt \ sT_DMSO_vs_untreated-down.txt \ -d$',' -o sT_DMSO_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ scr_Dox_vs_untreated-all.txt \ scr_Dox_vs_untreated-up.txt \ scr_Dox_vs_untreated-down.txt \ -d$',' -o scr_Dox_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ scr_DMSO_vs_untreated-all.txt \ scr_DMSO_vs_untreated-up.txt \ scr_DMSO_vs_untreated-down.txt \ -d$',' -o scr_DMSO_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_Dox_vs_untreated-all.txt \ sT_Dox_vs_untreated-up.txt \ sT_Dox_vs_untreated-down.txt \ -d$',' -o sT_Dox_vs_untreated.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_Dox_vs_sT_DMSO-all.txt \ sT_Dox_vs_sT_DMSO-up.txt \ sT_Dox_vs_sT_DMSO-down.txt \ -d$',' -o sT_Dox_vs_sT_DMSO.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_Dox_vs_scr_Dox-all.txt \ sT_Dox_vs_scr_Dox-up.txt \ sT_Dox_vs_scr_Dox-down.txt \ -d$',' -o sT_Dox_vs_scr_Dox.xls; ~/Tools/csv2xls-0.4/csv_to_xls.py \ sT_Dox_vs_scr_DMSO-all.txt \ sT_Dox_vs_scr_DMSO-up.txt \ sT_Dox_vs_scr_DMSO-down.txt \ -d$',' -o sT_Dox_vs_scr_DMSO.xls; # ------------------- volcano_plot ------------------- library(ggplot2) library(ggrepel) geness_res <- read.csv(file = paste("untreated_vs_parental_cells", "all.txt", sep="-"), row.names=1) external_gene_name <- rownames(geness_res) geness_res <- cbind(geness_res, external_gene_name) #top_g are from ids top_g <- c("hsa-miR-10b-5p","hsa-miR-1246","hsa-let-7a-5p","hsa-miR-182-5p","hsa-let-7f-5p","hsa-miR-1-3p","hsa-miR-375","hsa-miR-200c-3p","hsa-miR-30a-5p","hsa-miR-98-5p","hsa-miR-25-3p","hsa-miR-192-5p","hsa-miR-30c-5p","hsa-miR-1180-3p","hsa-let-7e-5p","hsa-miR-203a-3p","hsa-miR-625-3p","hsa-miR-146b-5p","hsa-miR-95-3p","hsa-miR-877-5p","hsa-miR-1307-3p","hsa-let-7c-5p","hsa-miR-361-5p","hsa-miR-30e-3p","hsa-miR-885-5p","hsa-miR-34a-5p","hsa-miR-93-5p","hsa-miR-5187-5p","hsa-miR-101-3p","hsa-miR-6850-5p","hsa-miR-103a-3p","hsa-miR-4511","hsa-miR-196a-5p","hsa-miR-1908-5p","hsa-miR-484","hsa-miR-92b-5p","hsa-miR-9-5p","hsa-miR-15b-5p","hsa-miR-30a-3p","hsa-miR-133b","hsa-miR-148a-3p","hsa-miR-1307-5p","hsa-miR-19b-3p","hsa-miR-6741-3p","hsa-miR-486-5p","hsa-miR-181a-5p","hsa-miR-342-5p","hsa-miR-873-3p","hsa-miR-324-5p","hsa-miR-769-5p","hsa-miR-328-3p","hsa-miR-301a-3p","hsa-miR-1224-5p","hsa-miR-671-5p","hsa-miR-652-3p","hsa-miR-1301-3p","hsa-miR-206","hsa-miR-889-3p","hsa-miR-197-3p","hsa-miR-217","hsa-miR-339-5p","hsa-miR-320c","hsa-miR-423-3p","hsa-miR-7706","hsa-miR-425-5p","hsa-miR-19a-3p","hsa-miR-149-5p","hsa-miR-361-3p","hsa-miR-4476","hsa-miR-186-5p","hsa-miR-342-3p","hsa-miR-708-3p","hsa-let-7b-5p","hsa-miR-17-5p","hsa-miR-532-3p","hsa-miR-1226-5p","hsa-miR-4677-3p","hsa-miR-3187-3p","hsa-miR-320a","hsa-miR-183-5p","hsa-miR-93-3p","hsa-miR-128-3p","hsa-miR-92a-1-5p","hsa-miR-501-5p","hsa-miR-454-3p","hsa-miR-760","hsa-miR-193b-3p","hsa-miR-200a-3p","hsa-miR-1290","hsa-miR-107","hsa-miR-331-3p","hsa-miR-148b-3p","hsa-miR-505-3p","hsa-miR-26b-5p","hsa-miR-130b-3p","hsa-miR-23b-3p","hsa-let-7g-5p","hsa-miR-188-5p","hsa-miR-432-5p","hsa-miR-190b","hsa-miR-1296-5p","hsa-miR-615-3p","hsa-miR-132-3p","hsa-miR-195-5p","hsa-miR-362-5p","hsa-miR-324-3p","hsa-miR-500a-3p","hsa-miR-151b","hsa-miR-92a-3p","hsa-miR-769-3p","hsa-miR-191-5p","hsa-miR-486-3p","hsa-miR-940","hsa-miR-449c-5p","hsa-miR-500a-5p","hsa-miR-22-3p","hsa-miR-183-3p","hsa-miR-181d-5p","hsa-miR-3200-3p","hsa-miR-1306-3p","hsa-miR-30c-2-3p","hsa-let-7b-3p","hsa-miR-1254","hsa-miR-7974","hsa-miR-216b-5p","hsa-miR-200b-5p","hsa-miR-1306-5p","hsa-miR-181b-5p","hsa-miR-133a-3p","hsa-miR-425-3p","hsa-miR-3934-5p","hsa-miR-421","hsa-miR-200b-3p","hsa-miR-18a-5p","hsa-miR-3605-5p","hsa-miR-210-3p","hsa-miR-193b-5p","hsa-miR-30b-5p","hsa-miR-190a-5p","hsa-miR-30e-5p","hsa-miR-106b-5p","hsa-miR-423-5p","hsa-mir-378c","hsa-miR-15a-5p","hsa-miR-92b-3p","hsa-miR-15b-3p","hsa-miR-148a-5p","hsa-miR-130b-5p","hsa-miR-181c-5p","hsa-miR-378e","hsa-miR-744-5p","hsa-miR-320b","hsa-miR-20a-5p","hsa-miR-885-3p","hsa-miR-339-3p","hsa-let-7i-5p","hsa-miR-181a-2-3p","hsa-miR-378i","hsa-miR-27b-3p","hsa-let-7a-3","hsa-miR-16-2-3p","hsa-miR-3615","hsa-miR-4510","hsa-miR-4492","hsa-miR-212-3p","hsa-let-7c","hsa-miR-660-5p","hsa-miR-25-5p","hsa-miR-16-5p","hsa-miR-141-3p","hsa-miR-30d-5p","hsa-let-7a-1","hsa-miR-151a-3p","hsa-let-7a-2","hsa-miR-30b-3p","hsa-miR-532-5p","hsa-miR-378d","hsa-let-7d-3p","hsa-miR-378c","hsa-miR-27a-3p","hsa-miR-378a-3p","hsa-miR-21-5p","hsa-miR-320d","hsa-miR-106b-3p","hsa-miR-320e","hsa-miR-196b-5p","hsa-miR-30d-3p","hsa-miR-4516","hsa-let-7b","hsa-miR-708-5p","hsa-miR-151a-5p|hsa-miR-151b","hsa-miR-6134","hsa-miR-106a-5p","hsa-miR-335-3p","hsa-miR-1269b","hsa-let-7d-5p","hsa-miR-139-3p","hsa-miR-218-5p","hsa-miR-6128","hsa-miR-215-5p","hsa-miR-26a-5p","hsa-miR-20b-5p","hsa-miR-24-3p","hsa-miR-330-3p","hsa-miR-941","hsa-miR-10a-5p","hsa-miR-1270","hsa-miR-345-5p","hsa-miR-140-3p","hsa-miR-7-5p","hsa-miR-577","hsa-let-7a-3p","hsa-miR-1269a","hsa-miR-1468-5p","hsa-miR-146a-5p") subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)) geness_res$Color <- "NS or log2FC < 2.0" geness_res$Color[geness_res$pvalue < 0.05] <- "P < 0.05" geness_res$Color[geness_res$padj < 0.05] <- "P-adj < 0.05" geness_res$Color[abs(geness_res$log2FoldChange) < 2.0] <- "NS or log2FC < 2.0" write.csv(geness_res, "untreated_vs_parental_cells_with_Category.csv") geness_res$invert_P <- (-log10(geness_res$pvalue)) * sign(geness_res$log2FoldChange) geness_res <- geness_res[, -1*ncol(geness_res)] png("volcano_plot_untreated_vs_parental_cells.png",width=1200, height=1400) #svg("untreated_vs_parental_cells.svg",width=12, height=14) ggplot(geness_res, aes(x = log2FoldChange, y = -log10(pvalue), color = Color, label = external_gene_name)) + geom_vline(xintercept = c(2.0, -2.0), lty = "dashed") + geom_hline(yintercept = -log10(0.05), lty = "dashed") + geom_point() + labs(x = "log2(FC)", y = "Significance, -log10(P)", color = "Significance") + scale_color_manual(values = c("P < 0.05"="orange","P-adj < 0.05"="red","NS or log2FC < 2.0"="darkgray"),guide = guide_legend(override.aes = list(size = 4))) + scale_y_continuous(expand = expansion(mult = c(0,0.05))) + geom_text_repel(data = subset(geness_res, external_gene_name %in% top_g & pvalue < 0.05 & (abs(geness_res$log2FoldChange) >= 2.0)), size = 4, point.padding = 0.15, color = "black", min.segment.length = .1, box.padding = .2, lwd = 2) + theme_bw(base_size = 16) + theme(legend.position = "bottom") dev.off() # ------------------ differentially_expressed_miRNAs_heatmap ----------------- # Batch Effect Removal Methods (Non-batch effect removal applied!) # prepare all_genes #rld <- rlogTransformation(dds) #mat <- assay(rld) #mm <- model.matrix(~replicates, colData(rld)) #mat <- limma::removeBatchEffect(mat, batch=rld$batch, design=mm) #assay(rld) <- mat RNASeq.NoCellLine <- assay(rld) #Manully defining miRNA for visualization for i in untreated_vs_parental_cells sT_Dox_vs_untreated sT_DMSO_vs_untreated scr_Dox_vs_untreated scr_DMSO_vs_untreated sT_Dox_vs_sT_DMSO sT_Dox_vs_scr_Dox sT_Dox_vs_scr_DMSO; do echo "cut -d',' -f1-1 ${i}-up.txt > ${i}-up.id"; echo "cut -d',' -f1-1 ${i}-down.txt > ${i}-down.id"; done #cat *.id | sort -u > ids ##add Gene_Id in the first line, delete the "" GOI <- read.csv("ids")$Gene_Id datamat = RNASeq.NoCellLine[GOI, ] # clustering the genes and draw heatmap #datamat <- datamat[,-1] #delete the sample "control MKL1" #datamat <- datamat[, 1:5] #parental_cells_1 parental_cells_2 parental_cells_3 untreated_1 untreated_2 scr_Dox_1 scr_Dox_2 scr_Dox_3 sT_DMSO_1 sT_DMSO_2 sT_DMSO_3 scr_DMSO_1 scr_DMSO_2 scr_DMSO_3 sT_Dox_1 sT_Dox_2 sT_Dox_3 --> #parental cells 1 parental cells 2 parental cells 3 untreated 1 untreated 2 scr control 1 scr control 2 scr control 3 DMSO control 1 DMSO control 2 DMSO control 3 scr DMSO control 1 scr DMSO control 2 scr DMSO control 3 sT knockdown 1 sT knockdown 2 sT knockdown 3 colnames(datamat)[1] <- "parental cells 1" colnames(datamat)[2] <- "parental cells 2" colnames(datamat)[3] <- "parental cells 3" colnames(datamat)[4] <- "untreated 1" colnames(datamat)[5] <- "untreated 2" colnames(datamat)[6] <- "scr Dox 1" colnames(datamat)[7] <- "scr Dox 2" colnames(datamat)[8] <- "scr Dox 3" colnames(datamat)[9] <- "sT DMSO 1" colnames(datamat)[10] <- "sT DMSO 2" colnames(datamat)[11] <- "sT DMSO 3" colnames(datamat)[12] <- "scr DMSO 1" colnames(datamat)[13] <- "scr DMSO 2" colnames(datamat)[14] <- "scr DMSO 3" colnames(datamat)[15] <- "sT Dox 1" colnames(datamat)[16] <- "sT Dox 2" colnames(datamat)[17] <- "sT Dox 3" write.csv(datamat, file ="differentially_expressed_miRNAs_heatmap.txt") write.xlsx(datamat, file = "differentially_expressed_miRNAs_heatmap.xlsx", rowNames = TRUE) #"ward.D"’, ‘"ward.D2"’,‘"single"’, ‘"complete"’, ‘"average"’ (= UPGMA), ‘"mcquitty"’(= WPGMA), ‘"median"’ (= WPGMC) or ‘"centroid"’ (= UPGMC) hr <- hclust(as.dist(1-cor(t(datamat), method="pearson")), method="complete") hc <- hclust(as.dist(1-cor(datamat, method="spearman")), method="complete") mycl = cutree(hr, h=max(hr$height)/1.1) mycol = c("YELLOW", "BLUE", "ORANGE", "CYAN", "GREEN", "MAGENTA", "GREY", "LIGHTCYAN", "RED", "PINK", "DARKORANGE", "MAROON", "LIGHTGREEN", "DARKBLUE", "DARKRED", "LIGHTBLUE", "DARKCYAN", "DARKGREEN", "DARKMAGENTA"); mycol = mycol[as.vector(mycl)] rownames(datamat) <- sub("\\|.*", "", rownames(datamat)) png("differentially_expressed_miRNAs_heatmap.png", width=1000, height=1400) heatmap.2(as.matrix(datamat), Rowv=as.dendrogram(hr), Colv=NA, dendrogram='row', labRow=row.names(datamat), scale='row', trace='none', col=bluered(75), RowSideColors=mycol, srtCol=30, lhei=c(1,8), cexRow=1.4, # Increase row label font size cexCol=1.7, # Increase column label font size margin=c(8, 12) ) dev.off() svg("differentially_expressed_miRNAs_heatmap.svg", width=12, height=16) heatmap.2(as.matrix(datamat), Rowv=as.dendrogram(hr), Colv=NA, dendrogram='row', labRow=row.names(datamat), scale='row', trace='none', col=bluered(75), RowSideColors=mycol, srtCol=30, lhei=c(1,8), cexRow=1.4, # Increase row label font size cexCol=1.7, # Increase column label font size margin=c(8, 12) ) dev.off() # mv differentially_expressed_miRNAs_heatmap.txt differentially_expressed_miRNAs_heatmap_MKL-1.txt # mv differentially_expressed_miRNAs_heatmap.xlsx differentially_expressed_miRNAs_heatmap_MKL-1.xlsx # mv differentially_expressed_miRNAs_heatmap.png differentially_expressed_miRNAs_heatmap_MKL-1.png # mv differentially_expressed_miRNAs_heatmap.svg differentially_expressed_miRNAs_heatmap_MKL-1.svg # mv distribution_heatmap.png distribution_heatmap_MKL-1.png # mv distribution_heatmap.svg distribution_heatmap_MKL-1.svg # mv volcano_plot_untreated_vs_parental_cells.png volcano_plot_untreated_vs_parental_cells_MKL-1.png