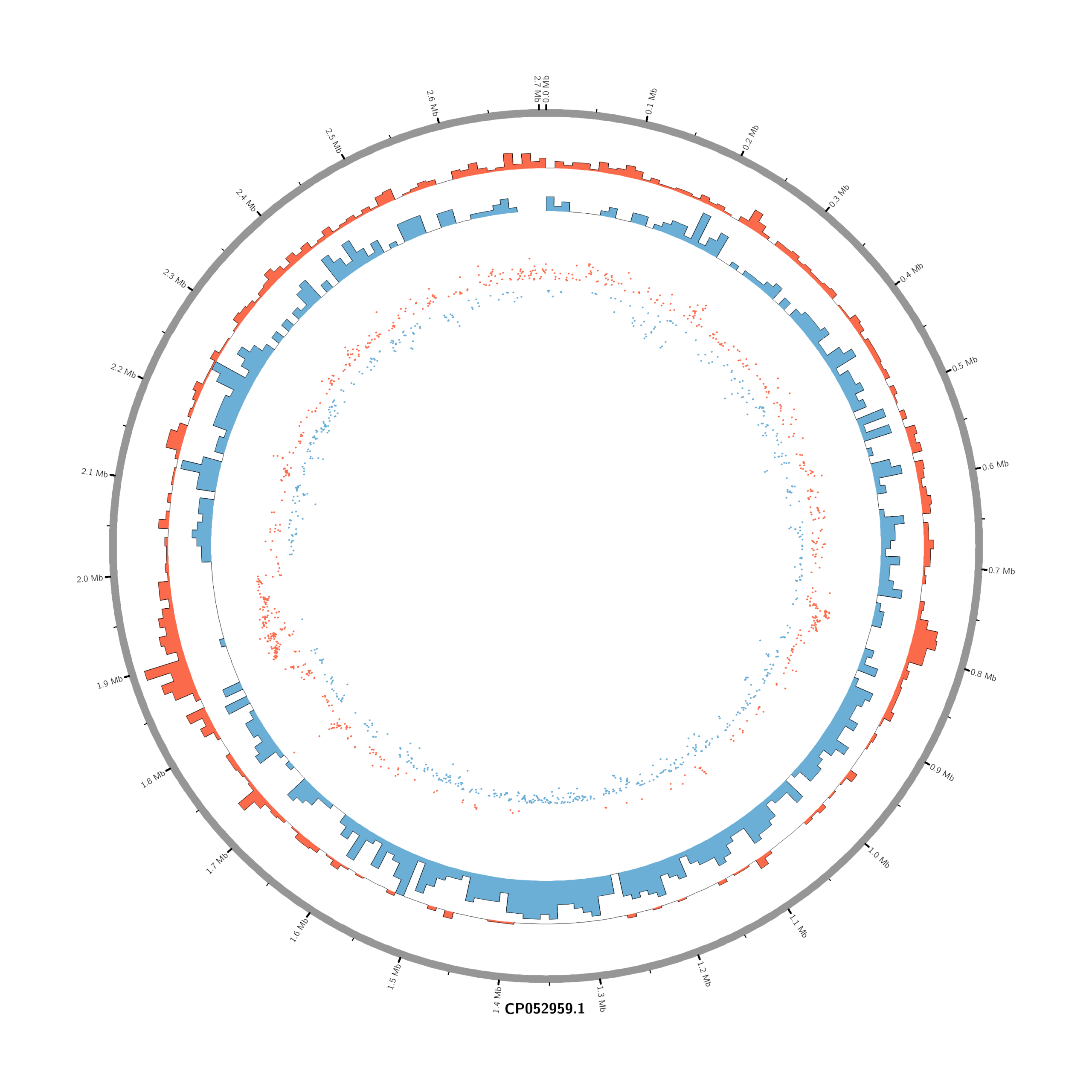
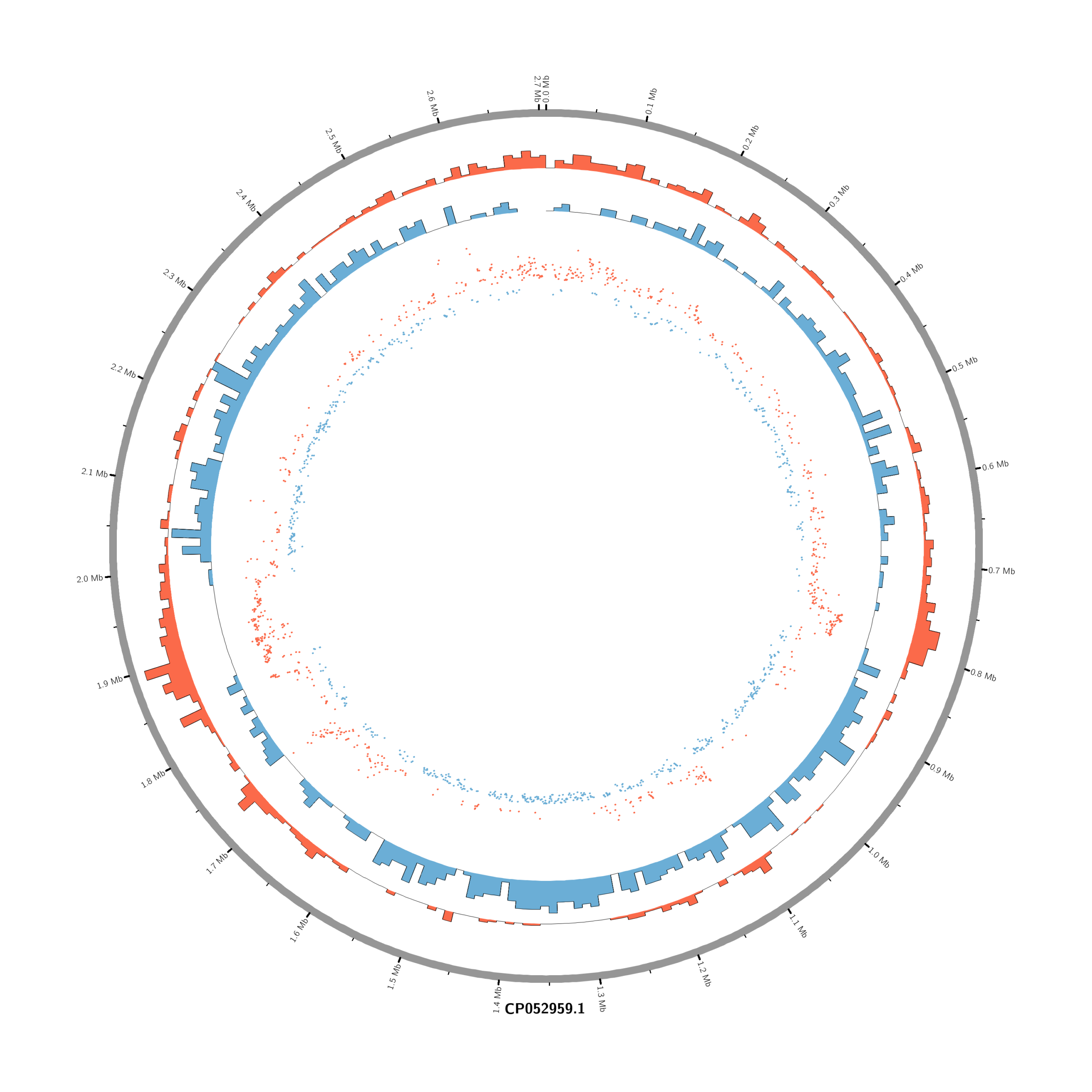
#Set the comparison condition in the R-script
comparison <- "Moxi_18h_vs_Untreated_18h"
# e.g.
# comparison <- "Mitomycin_18h_vs_Untreated_18h"
# comparison <- "Mitomycin_8h_vs_Untreated_8h"
# comparison <- "Moxi_8h_vs_Untreated_8h"
# comparison <- "Mitomycin_4h_vs_Untreated_4h"
# comparison <- "Moxi_4h_vs_Untreated_4h"
(r_env) Rscript make_circos_from_deseq.R
(circos-env) cd circos_Moxi_18h_vs_Untreated_18h
(circos-env) touch karyotype.txt (see the step2)
(circos-env) touch circos.conf (see the step3)
cp circos_Moxi_18h_vs_Untreated_18h/karyotype.txt circos_Moxi_8h_vs_Untreated_8h/
cp circos_Moxi_18h_vs_Untreated_18h/karyotype.txt circos_Moxi_4h_vs_Untreated_4h/
cp circos_Moxi_18h_vs_Untreated_18h/karyotype.txt circos_Mitomycin_18h_vs_Untreated_18h/
cp circos_Moxi_18h_vs_Untreated_18h/karyotype.txt circos_Mitomycin_8h_vs_Untreated_8h/
cp circos_Moxi_18h_vs_Untreated_18h/karyotype.txt circos_Mitomycin_4h_vs_Untreated_4h/
cp circos_Moxi_18h_vs_Untreated_18h/circos.conf circos_Moxi_8h_vs_Untreated_8h/
cp circos_Moxi_18h_vs_Untreated_18h/circos.conf circos_Moxi_4h_vs_Untreated_4h/
cp circos_Moxi_18h_vs_Untreated_18h/circos.conf circos_Mitomycin_18h_vs_Untreated_18h/
cp circos_Moxi_18h_vs_Untreated_18h/circos.conf circos_Mitomycin_8h_vs_Untreated_8h/
cp circos_Moxi_18h_vs_Untreated_18h/circos.conf circos_Mitomycin_4h_vs_Untreated_4h/
(circos-env) circos -conf circos.conf
#or (circos-env) /home/jhuang/mambaforge/envs/circos-env/bin/circos -conf /mnt/md1/DATA/Data_JuliaFuchs_RNAseq_2025/results/star_salmon/degenes/circos/circos.conf
#(circos-env) jhuang@WS-2290C:/mnt/md1/DATA/Data_JuliaFuchs_RNAseq_2025/results/star_salmon/degenes$ find . -name "*circos.png"
mv ./circos_Moxi_4h_vs_Untreated_4h/circos.png circos_Moxi_4h_vs_Untreated_4h.png
mv ./circos_Moxi_18h_vs_Untreated_18h/circos.png circos_Moxi_18h_vs_Untreated_18h.png
mv ./circos_Moxi_8h_vs_Untreated_8h/circos.png circos_Moxi_8h_vs_Untreated_8h.png
mv ./circos_Mitomycin_18h_vs_Untreated_18h/circos.png circos_Mitomycin_18h_vs_Untreated_18h.png
mv ./circos_Mitomycin_8h_vs_Untreated_8h/circos.png circos_Mitomycin_8h_vs_Untreated_8h.png
mv ./circos_Mitomycin_4h_vs_Untreated_4h/circos.png circos_Mitomycin_4h_vs_Untreated_4h.png
- touch karyotype.txt
chr - CP052959.1 CP052959.1 0 2706926 grey- touch circos.conf
karyotype = karyotype.txt
<ideogram>
<spacing>
default = 0.005r
</spacing>
radius = 0.80r
thickness = 20p
fill = yes
show_label = yes
label_radius = 1.05r
label_size = 30p
label_font = bold
label_parallel = yes
</ideogram>
# --- Wichtig: Schalter auf Top-Level, NICHT im
<ticks>-Block ---
show_ticks = yes
show_tick_labels = yes
<ticks>
# Starte direkt an der äußeren Ideogramm-Kante
radius = dims(ideogram,radius_outer)
orientation = out # Ticks nach außen zeichnen (oder 'in' für nach innen)
color = black
thickness = 2p
size = 8p
# kleine Ticks alle 100 kb, ohne Label
<tick>
spacing = 50000
size = 8p
thickness = 3p
color = black
show_label = no
</tick>
# große Ticks alle 500 kb, mit Label in Mb
<tick>
spacing = 100000
size = 12p
thickness = 5p
color = black
show_label = yes
label_size = 18p
label_offset = 6p
multiplier = 0.000001 # in Mb
format = %.1f
suffix = " Mb"
</tick>
</ticks>
<plots>
# Density of up-regulated genes
<plot>
show = yes
type = histogram
file = data/density_up.txt
r0 = 0.88r
r1 = 0.78r
fill_color = red
thickness = 1p
</plot>
# Density of down-regulated genes
<plot>
show = yes
type = histogram
file = data/density_down.txt
r0 = 0.78r
r1 = 0.68r
fill_color = blue
thickness = 1p
</plot>
# Scatter of individual significantly DE genes
<plot>
show = yes
type = scatter
file = data/genes_scatter.txt
r0 = 0.46r
r1 = 0.76r
glyph = circle
glyph_size = 5p
stroke_thickness = 0
min = -15
max = 15
<rules>
<rule>
condition = var(value) > 0
color = red
</rule>
<rule>
condition = var(value) < 0
color = blue
</rule>
</rules>
</plot>
</plots>
<image>
<<include etc/image.conf>>
</image>
<<include etc/colors_fonts_patterns.conf>>
<<include etc/housekeeping.conf>>-
Rscript make_circos_from_deseq.R
############################################################ # make_circos_from_deseq.R # # - Read DESeq2 results (annotated CSV) # - Read BED annotation # - Merge by GeneID # - Classify genes (up / down / ns) # - Create Circos input files: # * genes_scatter.txt # * density_up.txt # * density_down.txt # - Create annotated versions of the above # - Export annotated tables into one Excel workbook ############################################################ ############################### # 0. Single parameter to set ############################### # Just change this line for each comparison: #comparison <- "Moxi_18h_vs_Untreated_18h" # e.g. #comparison <- "Mitomycin_18h_vs_Untreated_18h" #comparison <- "Mitomycin_8h_vs_Untreated_8h" #comparison <- "Moxi_8h_vs_Untreated_8h" #comparison <- "Mitomycin_4h_vs_Untreated_4h" comparison <- "Moxi_4h_vs_Untreated_4h" ############################### # 0. Derived settings ############################### # DESeq2 result file (annotated) deseq_file <- paste0(comparison, "-all_annotated.csv") # BED file with gene coordinates (constant for this genome) bed_file <- "CP052959_m.bed" # Create a filesystem-friendly directory name from comparison safe_comp <- gsub("[^A-Za-z0-9_]+", "_", comparison) # Output directory for Circos files (e.g. circos_Moxi_18h_vs_Untreated_18h) out_dir <- paste0("circos_", safe_comp) # Thresholds for significance padj_cutoff <- 0.05 lfc_cutoff <- 1 # |log2FC| >= 1 # Bin size for density in bp (e.g. 10000 = 10 kb) bin_size <- 10000 # Comparison label for Excel / plots (human-readable) comparison_label <- comparison options(scipen = 999) # turn off scientific notation ############################### # 1. Setup & packages ############################### if (!dir.exists(out_dir)) dir.create(out_dir, showWarnings = FALSE) data_dir <- file.path(out_dir, "data") if (!dir.exists(data_dir)) dir.create(data_dir, showWarnings = FALSE) # openxlsx for Excel export if (!requireNamespace("openxlsx", quietly = TRUE)) { stop("Package 'openxlsx' is required. Please install it with install.packages('openxlsx').") } library(openxlsx) ############################### # 2. Read data ############################### message("Reading DESeq2 results from: ", deseq_file) deseq <- read.csv(deseq_file, stringsAsFactors = FALSE) # Check required columns in DESeq2 table required_cols <- c("GeneID", "log2FoldChange", "padj") if (!all(required_cols %in% colnames(deseq))) { stop("DESeq2 table must contain columns: ", paste(required_cols, collapse = ", ")) } message("Reading BED annotation from: ", bed_file) bed_cols <- c("chr","start","end","gene_id","score", "strand","thickStart","thickEnd", "itemRgb","blockCount","blockSizes","blockStarts") annot <- read.table( bed_file, header = FALSE, sep = "\t", stringsAsFactors = FALSE, col.names = bed_cols ) ############################### # 3. Merge DESeq2 + annotation ############################### # Match DESeq2 GeneID (e.g. "gene-HJI06_09365") to BED gene_id merged <- merge( deseq, annot[, c("chr", "start", "end", "gene_id")], by.x = "GeneID", by.y = "gene_id" ) if (nrow(merged) == 0) { stop("No overlap between DESeq2 GeneID and BED gene_id.") } # Midpoint of each gene merged$mid <- round((merged$start + merged$end) / 2) ############################### # 4. Classify genes ############################### merged$regulation <- "ns" merged$regulation[merged$padj < padj_cutoff & merged$log2FoldChange >= lfc_cutoff] <- "up" merged$regulation[merged$padj < padj_cutoff & merged$log2FoldChange <= -lfc_cutoff] <- "down" table_reg <- table(merged$regulation) message("Regulation counts: ", paste(names(table_reg), table_reg, collapse = " | ")) ############################### # 5. Scatter files (per-gene) ############################### scatter <- merged[merged$regulation != "ns", ] # Circos scatter input: chr start end value scatter_file <- file.path(data_dir, "genes_scatter.txt") scatter_out <- scatter[, c("chr", "mid", "mid", "log2FoldChange")] write.table(scatter_out, scatter_file, quote = FALSE, sep = "\t", row.names = FALSE, col.names = FALSE) message("Written Circos scatter file: ", scatter_file) # Annotated scatter for Excel / inspection scatter_annot <- scatter[, c( "chr", "mid", # start "mid", # end "log2FoldChange", "GeneID", "padj", "regulation" )] colnames(scatter_annot)[1:4] <- c("chr", "start", "end", "log2FoldChange") scatter_annot_file <- file.path(data_dir, "genes_scatter_annotated.tsv") write.table(scatter_annot, scatter_annot_file, quote = FALSE, sep = "\t", row.names = FALSE, col.names = TRUE) message("Written annotated scatter file: ", scatter_annot_file) ############################### # 6. Density function (bins) ############################### bin_chr <- function(df_chr, bin_size, direction = c("up", "down")) { direction <- match.arg(direction) chr_name <- df_chr$chr[1] max_pos <- max(df_chr$mid) # number of bins n_bins <- ceiling((max_pos + 1) / bin_size) starts <- seq(0, by = bin_size, length.out = n_bins) ends <- starts + bin_size # init counts & gene lists counts <- integer(n_bins) gene_list <- vector("list", n_bins) df_dir <- df_chr[df_chr$regulation == direction, ] if (nrow(df_dir) > 0) { # bin index for each gene bin_index <- floor(df_dir$mid / bin_size) + 1 bin_index[bin_index < 1] <- 1 bin_index[bin_index > n_bins] <- n_bins # accumulate counts and GeneIDs for (i in seq_len(nrow(df_dir))) { idx <- bin_index[i] counts[idx] <- counts[idx] + 1L gene_list[[idx]] <- c(gene_list[[idx]], df_dir$GeneID[i]) } } gene_ids <- vapply( gene_list, function(x) { if (length(x) == 0) "" else paste(unique(x), collapse = ";") }, character(1) ) data.frame( chr = chr_name, start = as.integer(starts), end = as.integer(ends), value = as.integer(counts), gene_ids = gene_ids, stringsAsFactors = FALSE ) } ############################### # 7. Density up/down for all chromosomes ############################### chr_list <- split(merged, merged$chr) density_up_list <- lapply(chr_list, bin_chr, bin_size = bin_size, direction = "up") density_down_list <- lapply(chr_list, bin_size = bin_size, FUN = bin_chr, direction = "down") density_up <- do.call(rbind, density_up_list) density_down <- do.call(rbind, density_down_list) # Plain Circos input (no gene_ids) density_up_file <- file.path(data_dir, "density_up.txt") density_down_file <- file.path(data_dir, "density_down.txt") write.table(density_up[, c("chr", "start", "end", "value")], density_up_file, quote = FALSE, sep = "\t", row.names = FALSE, col.names = FALSE) write.table(density_down[, c("chr", "start", "end", "value")], density_down_file, quote = FALSE, sep = "\t", row.names = FALSE, col.names = FALSE) message("Written Circos density files: ", density_up_file, " and ", density_down_file) # Annotated density files (with gene_ids) density_up_annot_file <- file.path(data_dir, "density_up_annotated.tsv") density_down_annot_file <- file.path(data_dir, "density_down_annotated.tsv") write.table(density_up, density_up_annot_file, quote = FALSE, sep = "\t", row.names = FALSE, col.names = TRUE) write.table(density_down, density_down_annot_file, quote = FALSE, sep = "\t", row.names = FALSE, col.names = TRUE) message("Written annotated density files: ", density_up_annot_file, " and ", density_down_annot_file) ############################### # 8. Export annotated tables to Excel ############################### excel_file <- file.path( out_dir, paste0("circos_annotations_", comparison_label, ".xlsx") ) wb <- createWorkbook() addWorksheet(wb, "scatter_points") writeData(wb, "scatter_points", scatter_annot) addWorksheet(wb, "density_up") writeData(wb, "density_up", density_up) addWorksheet(wb, "density_down") writeData(wb, "density_down", density_down) saveWorkbook(wb, excel_file, overwrite = TRUE) message("Excel workbook written to: ", excel_file) message("Done.")