This post is a standalone, reproducible record of the bacterial WGS pipeline I used (example sample: AN6). I’m keeping all command lines (as-run) so you can reuse the workflow for future projects. Wherever you see absolute paths, replace them with your own.
0) Prerequisites (what you need installed)
- Nextflow
- Docker (for nf-core/bacass
-profile docker) - Conda/Mamba
- CLI tools used later:
fastqc,spades.py,shovill,pigz,awk,seqkit,fastANI, plus R (for plotting), and the tools required by the provided scripts.
1) Download KmerFinder database
# Download the kmerfinder database: https://www.genomicepidemiology.org/services/ --> https://cge.food.dtu.dk/services/KmerFinder/ --> https://cge.food.dtu.dk/services/KmerFinder/etc/kmerfinder_db.tar.gz
# Download 20190108_kmerfinder_stable_dirs.tar.gz from https://zenodo.org/records/134470562) Run nf-core/bacass (Nextflow)
#--kmerfinderdb /path/to/kmerfinder/bacteria.tar.gz
#--kmerfinderdb /mnt/nvme1n1p1/REFs/kmerfinder_db.tar.gz
#--kmerfinderdb /mnt/nvme1n1p1/REFs/20190108_kmerfinder_stable_dirs.tar.gz
nextflow run nf-core/bacass -r 2.5.0 -profile docker \
--input samplesheet.tsv \
--outdir bacass_out \
--assembly_type long \
--kraken2db /mnt/nvme1n1p1/REFs/k2_standard_08_GB_20251015.tar.gz \
--kmerfinderdb /mnt/nvme1n1p1/REFs/kmerfinder/bacteria/ \
-resume
#SAVE bacass_out/Kmerfinder/kmerfinder_summary.csv to bacass_out/Kmerfinder/An6/An6_kmerfinder_results.xlsx3) Assembly (AN6 example)
3.1 Link raw reads + run FastQC
ln -s ../X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz An6_R1.fastq.gz
ln -s ../X101SC25116512-Z01-J002/01.RawData/An6/An6_2.fq.gz An6_R2.fastq.gz
mkdir fastqc_out
fastqc -t 4 raw_data/* -o fastqc_out/
mamba activate /home/jhuang/miniconda3/envs/bengal3_ac33.2 Trimming decision notes (kept as recorded)
For the AN6 data, it’s not better to run Trimmomatic first in most cases (adapters OK; per-tile failures are instrument/tile related and not “fixed” by trimming).
* **Adapters:** FastQC shows **Adapter Content = PASS** for both R1/R2. * **Overrepresented sequences:** none detected. * **Per-tile sequence quality:** **FAIL** (this is usually an instrument/tile effect; trimming adapters won’t “fix” it).Shovill: avoid pre-trimming (default read trimming already included unless disabled). SPAdes: trimming optional; try raw first, then trimmed if needed.
3.3 If you do need Trimmomatic (command kept)
# Paired-end trimming with Trimmomatic (Illumina-style)
# Adjust TRIMMOMATIC_JAR and ADAPTERS paths to your install.
TRIMMOMATIC_JAR=/path/to/trimmomatic.jar
ADAPTERS=/path/to/Trimmomatic/adapters/TruSeq3-PE.fa
java -jar "$TRIMMOMATIC_JAR" PE -threads 16 -phred33 \
An6_R1.fastq.gz An6_R2.fastq.gz \
An6_R1.trim.paired.fastq.gz An6_R1.trim.unpaired.fastq.gz \
An6_R2.trim.paired.fastq.gz An6_R2.trim.unpaired.fastq.gz \
ILLUMINACLIP:"$ADAPTERS":2:30:10 \
LEADING:3 TRAILING:3 \
SLIDINGWINDOW:4:20 \
MINLEN:50What you feed into SPAdes/Shovill afterward:
-
Use the paired outputs:
An6_R1.trim.paired.fastq.gzAn6_R2.trim.paired.fastq.gz
- Optional: you can include unpaired reads in SPAdes, but many people skip them for isolate assemblies unless coverage is low.
If you want, I can also give the matching SPAdes command that includes unpaired reads (or the cleanest approach that ignores them).
spades.py \
-1 raw_data/An6_R1.fastq.gz \
-2 raw_data/An6_R2.fastq.gz \
--isolate \
-t 32 -m 250 \
-o spades_out
spades.py \
-1 raw_data/An6_R1.fastq.gz \
-2 raw_data/An6_R2.fastq.gz \
--careful \
-t 32 -m 250 \
-o spades_out_carefulShovill (CHOSEN; default does read trimming unless you disable it):
shovill \
--R1 raw_data/An6_R1.fastq.gz \
--R2 raw_data/An6_R2.fastq.gz \
--outdir shovill_out \
--cpus 32 --ram 250 \
--depth 100If you want to keep reads completely untrimmed in Shovill, add --noreadtrim.
4) Genome annotation — BV-BRC ComprehensiveGenomeAnalysis
* Use: https://www.bv-brc.org/app/ComprehensiveGenomeAnalysis
* Input: scaffolded results from bacass
* Purpose: comprehensive overview + annotation of the genome assembly.5) Table 1 — summary of sequence data + genome features (env: gunc_env)
5.1 Environment prep + pipeline run (kept)
# Prepare environment and run the Table 1 (Summary of sequence data and genome features (env: gunc_env)) pipeline:
# activate the env that has openpyxl
mamba activate gunc_env
mamba install -n gunc_env -c conda-forge openpyxl -y
mamba deactivate
# STEP_1
ENV_NAME=gunc_env \
SAMPLE=AN6 \
ASM=shovill_out/contigs.fa \
R1=./X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz \
R2=./X101SC25116512-Z01-J002/01.RawData/An6/An6_2.fq.gz \
./make_table1_pe.sh
# STEP_2
python export_table1_stats_to_excel_py36_compat.py \
--workdir table1_AN6_work \
--out Comprehensive_AN6.xlsx \
--max-rows 200000 \
--sample AN65.2 Manual calculations (kept)
#Manually For the items “Total number of reads sequenced” and “Mean read length (bp)”:
#Total number of reads sequenced 9,127,297 × 2
#Coverage depth (sequencing depth) 589.4×
pigz -dc X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz | awk 'END{print NR/4}'
seqkit stats X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz
#file format type num_seqs sum_len min_len avg_len max_len
#X101SC25116512-Z01-J002/01.RawData/An6/An6_1.fq.gz FASTQ DNA 15,929,405 2,389,410,750 150 150 1505.3 Example metrics table snapshot (kept)
Metricsa Value
Genome size (bp) 3,012,410
Contig count (>= 500 bp) 41
Total number of reads sequenced 15,929,405 × 2
Coverage depth (sequencing depth) 1454.3×
Coarse consistency (%) 99.67
Fine consistency (%) 94.50
Completeness (%) 99.73
Contamination (%) 0.21
Contigs N50 (bp) 169,757
Contigs L50 4
Guanine-cytosine content (%) 41.14
Number of coding sequences (CDSs) 2,938
Number of tRNAs 69
Number of rRNAs 36) AMR / virulence screening (ABRicate workflows)
cp shovill_out/contigs.fa AN6.fasta
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ASM=AN6.fasta SAMPLE=AN6 THREADS=32 ./run_resistome_virulome_dedup.sh #Default MINID=90 MINCOV=60
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ASM=AN6.fasta SAMPLE=AN6 MINID=80 MINCOV=60 ./run_resistome_virulome_dedup.sh # 0 0 0 0
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ASM=AN6.fasta SAMPLE=AN6 MINID=70 MINCOV=50 ./run_resistome_virulome_dedup.sh # 5 5 0 4
#Sanity checks on ABRicate outputs
grep -vc '^#' resistome_virulence_AN6/raw/AN6.megares.tab
grep -vc '^#' resistome_virulence_AN6/raw/AN6.card.tab
grep -vc '^#' resistome_virulence_AN6/raw/AN6.resfinder.tab
grep -vc '^#' resistome_virulence_AN6/raw/AN6.vfdb.tab
#!!!!!! DEBUG_TOMORROW: why using 'MINID=70 MINCOV=50' didn't return the 5504?
#Dedup tables / “one per gene” mode
rm Resistome_Virulence_An6.xlsx
chmod +x run_abricate_resistome_virulome_one_per_gene.sh
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 \
ASM=AN6.fasta \
SAMPLE=AN6 \
OUTDIR=resistome_virulence_AN6 \
MINID=70 MINCOV=50 \
THREADS=32 \
./run_abricate_resistome_virulome_one_per_gene.sh
cd resistome_virulence_AN6
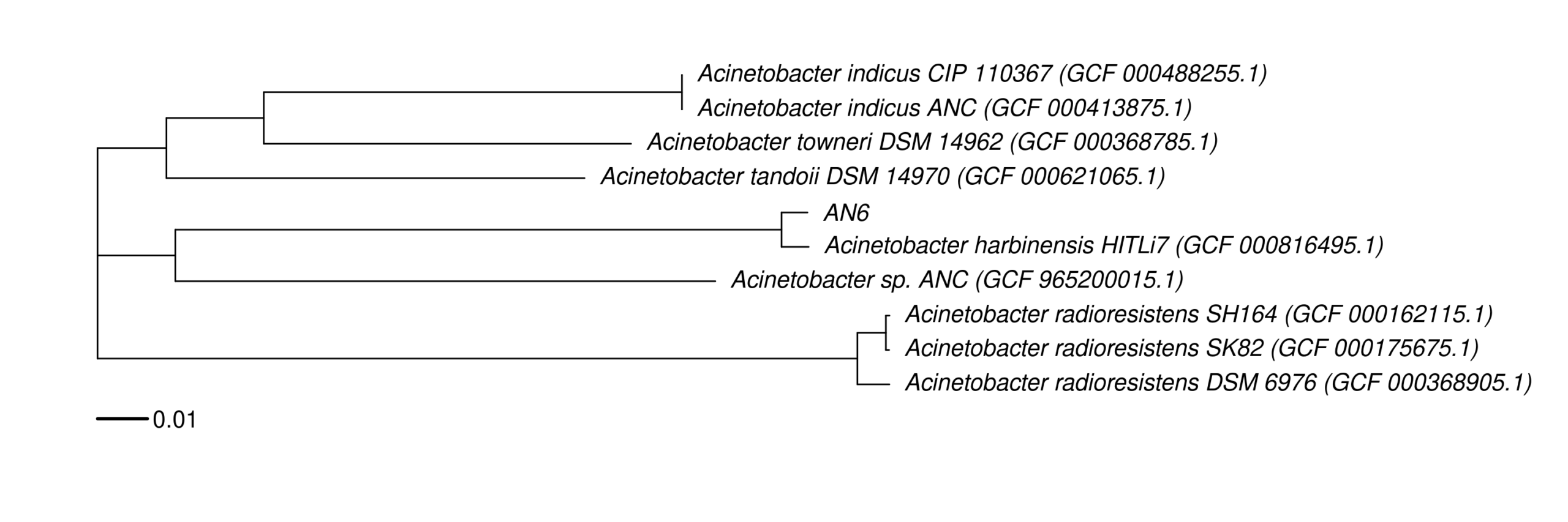
python3 -c 'import pandas as pd; from pathlib import Path; files=["Table_AMR_genes_dedup.tsv","Table_AMR_genes_one_per_gene.tsv","Table_Virulence_VFDB_dedup.tsv","Table_DB_hit_counts.tsv"]; out="AN6_resistome_virulence.xlsx"; w=pd.ExcelWriter(out, engine="openpyxl"); [pd.read_csv(f, sep="\t").to_excel(w, sheet_name=Path(f).stem[:31], index=False) for f in files]; w.close(); print(out)'7) Core-genome phylogeny (NCBI + Roary + RAxML-NG + R plotting)
#Generate targets.tsv from ./bvbrc_out/Acinetobacter_harbinensis_AN6/FullGenomeReport.html.
export NCBI_EMAIL="xxx@yyy.de"
./resolve_best_assemblies_entrez.py targets.tsv resolved_accessions.tsv
#[OK] Acinetobacter_harbinensis_HITLi7 -> GCF_000816495.1 (Scaffold)
#[OK] Acinetobacter_sp._ANC -> GCF_965200015.1 (Complete Genome)
#[OK] Acinetobacter_sp._TTH0-4 -> GCF_965200015.1 (Complete Genome)
#[OK] Acinetobacter_tandoii_DSM_14970 -> GCF_000621065.1 (Scaffold)
#[OK] Acinetobacter_towneri_DSM_14962 -> GCF_000368785.1 (Scaffold)
#[OK] Acinetobacter_radioresistens_SH164 -> GCF_000162115.1 (Scaffold)
#[OK] Acinetobacter_radioresistens_SK82 -> GCF_000175675.1 (Contig)
#[OK] Acinetobacter_radioresistens_DSM_6976 -> GCF_000368905.1 (Scaffold)
#[OK] Acinetobacter_indicus_ANC -> GCF_000413875.1 (Scaffold)
#[OK] Acinetobacter_indicus_CIP_110367 -> GCF_000488255.1 (Scaffold)
#NOTE the env bengal3_ac3 don’t have the following R package, using r_env for the plot-step → RUN TWICE, first bengal3_ac3, then run build_wgs_tree_fig3B.sh plot-only.
#ADAPT the params EXTRA_ASSEMBLIES (could stay as empty), and AN6.fasta as REF_FASTA
conda activate /home/jhuang/miniconda3/envs/bengal3_ac3
export NCBI_EMAIL="xxx@yyy.de"
ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3 ./build_wgs_tree_fig3B.sh
# (Optional) if want to delete some leaves from the tree, remove from inputs so Roary cannot include it
for id in "GCF_002291425.1" "GCF_047901425.1" "GCF_004342245.1" "GCA_032062225.1"; do
rm -f work_wgs_tree/gffs/${id}.gff
rm -f work_wgs_tree/fastas/${id}.fna
rm -rf work_wgs_tree/prokka/${id}
rm -rf work_wgs_tree/genomes_ncbi/${id}
# remove from accession list so it won't come back
awk -F'\t' 'NR==1 || $2!="${id}"' work_wgs_tree/meta/accessions.tsv > work_wgs_tree/meta/accessions.tsv.tmp \
&& mv work_wgs_tree/meta/accessions.tsv.tmp work_wgs_tree/meta/accessions.tsv
done
./build_wgs_tree_fig3B.sh
#Wrote: work_wgs_tree/plot/labels.tsv
#Error: package or namespace load failed for ‘ggtree’ in loadNamespace(j <- i[[1L]], c(lib.loc, .libPaths()), versionCheck = vI[[j]]):
#there is no package called ‘aplot’
#Execution halted --> Using env r_env instead (see below)!
# Run this to regenerate labels.tsv
bash regenerate_labels.sh
# Regenerate the plot --> ERROR --> Using Rscript instead (see below)!
ENV_NAME=/home/jhuang/mambaforge/envs/r_env ./build_wgs_tree_fig3B.sh plot-only
#-->Error in as.hclust.phylo(tr) : the tree is not ultrametric
# 8) Manual correct the display name in work_wgs_tree/plot/labels.tsv
#sample display
#GCF_000816495.1 Acinetobacter harbinensis HITLi7 (GCF_000816495.1)
#GCF_965200015.1 Acinetobacter sp. ANC (GCF_965200015.1)
#GCF_000621065.1 Acinetobacter tandoii DSM 14970 (GCF_000621065.1)
#GCF_000368785.1 Acinetobacter towneri DSM 14962 (GCF_000368785.1)
#GCF_000162115.1 Acinetobacter radioresistens SH164 (GCF_000162115.1)
#GCF_000175675.1 Acinetobacter radioresistens SK82 (GCF_000175675.1)
#GCF_000368905.1 Acinetobacter radioresistens DSM 6976 (GCF_000368905.1)
#GCF_000413875.1 Acinetobacter indicus ANC (GCF_000413875.1)
#GCF_000488255.1 Acinetobacter indicus CIP 110367 (GCF_000488255.1)
#REF AN6
# 9) Rerun only the plot step uisng plot_tree_v4.R
Rscript ./plot_tree_v4.R \
work_wgs_tree/raxmlng/core.raxml.support \
work_wgs_tree/plot/labels.tsv \
6 \
work_wgs_tree/plot/core_tree.pdf \
work_wgs_tree/plot/core_tree.png8) ANI confirmation (fastANI loop)
mamba activate /home/jhuang/miniconda3/envs/bengal3_ac3
for id in GCF_000621065.1.fna GCF_000368785.1.fna GCF_000175675.1.fna GCF_000368905.1.fna GCF_000816495.1.fna GCF_965200015.1.fna GCF_000488255.1.fna GCF_000413875.1.fna GCF_000162115.1.fna; do
fastANI -q AN6.fasta -r ./work_wgs_tree/fastas/${id} -o fastANI_AN6_vs_${id}.txt
done
# Alternatively, we can use the script run_fastani_batch_verbose.sh.9) Contig-to-reference mapping (how many contigs map?)
In total, we obtained 41 contigs >500 nt. Of these, 36 contigs were scaffolded with Multi-CSAR v1.1 into three chromosomal scaffolds:
- SCF_1: 1,773,912 bp
- SCF_2: 1,197,749 bp
- SCF_3: 23,925 bp Total: 2,995,586 bp
The remaining five contigs (contig00026/32/33/37/39) could not be scaffolded. Their partial BLASTn matches to both plasmid and chromosomal sequences suggest shared mobile elements, but do not confirm circular plasmids. A sequence/assembly summary was exported to Excel (Summary_AN6.xlsx), including read yield/read-length statistics and key assembly/QC metrics (genome size, contigs/scaffolds, N50, GC%, completeness, contamination).
Complete scripts (as attached)
Below are the full scripts exactly as provided, including plot_tree_v4.R.
make_table1_pe.sh
#!/usr/bin/env bash
set -Eeuo pipefail
# =========================
# User config
ENV_NAME="${ENV_NAME:-checkm_env2}"
# If you have Illumina paired-end, set R1/R2 (recommended)
R1="${R1:-}"
R2="${R2:-}"
# If you have single-end/ONT-like reads, set READS instead (legacy mode)
READS="${READS:-}"
ASM="${ASM:-shovill_out/contigs.fa}"
SAMPLE="${SAMPLE:-An6}"
THREADS="${THREADS:-32}"
OUT_TSV="${OUT_TSV:-Table1_${SAMPLE}.tsv}"
WORKDIR="${WORKDIR:-table1_${SAMPLE}_work}"
LOGDIR="${LOGDIR:-${WORKDIR}/logs}"
LOGFILE="${LOGFILE:-${LOGDIR}/run_$(date +%F_%H%M%S).log}"
AUTO_INSTALL="${AUTO_INSTALL:-1}" # 1=install missing tools in ENV_NAME
GUNC_DB_KIND="${GUNC_DB_KIND:-progenomes}" # progenomes or gtdb
# =========================
mkdir -p "${LOGDIR}"
exec > >(tee -a "${LOGFILE}") 2>&1
ts(){ date +"%F %T"; }
log(){ echo "[$(ts)] $*"; }
on_err() {
local ec=$?
log "ERROR: failed (exit=${ec}) at line ${BASH_LINENO[0]}: ${BASH_COMMAND}"
log "Logfile: ${LOGFILE}"
exit "${ec}"
}
trap on_err ERR
# print every command
set -x
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
pick_pm() {
if need_cmd mamba; then echo "mamba"
elif need_cmd conda; then echo "conda"
else
log "ERROR: neither mamba nor conda found in PATH"
exit 1
fi
}
activate_env() {
if ! need_cmd conda; then
log "ERROR: conda not found; cannot activate env"
exit 1
fi
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
conda activate "${ENV_NAME}"
}
ensure_env_exists() {
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
if ! conda env list | awk '{print $1}' | grep -qx "${ENV_NAME}"; then
log "ERROR: env ${ENV_NAME} not found. Create it first."
exit 1
fi
}
install_pkgs_in_env() {
local pm="$1"; shift
local pkgs=("$@")
log "Installing into env ${ENV_NAME}: ${pkgs[*]}"
"${pm}" install -n "${ENV_NAME}" -c bioconda -c conda-forge -y "${pkgs[@]}"
}
pick_quast_cmd() {
if need_cmd quast; then echo "quast"
elif need_cmd quast.py; then echo "quast.py"
else echo ""
fi
}
# tool->package mapping (install missing ones)
declare -A TOOL2PKG=(
[quast]="quast"
[minimap2]="minimap2"
[samtools]="samtools"
[mosdepth]="mosdepth"
[checkm]="checkm-genome=1.1.3"
[gunc]="gunc"
[python]="python"
)
# =========================
# Detect mode (PE vs single)
MODE=""
if [[ -n "${R1}" || -n "${R2}" ]]; then
[[ -n "${R1}" && -n "${R2}" ]] || { log "ERROR: Provide both R1 and R2."; exit 1; }
MODE="PE"
elif [[ -n "${READS}" ]]; then
MODE="SINGLE"
else
log "ERROR: Provide either (R1+R2) OR READS."
exit 1
fi
# =========================
# Start
log "Start: Table 1 generation (reuse env=${ENV_NAME})"
log "Assembly: ${ASM}"
log "Sample: ${SAMPLE}"
log "Threads: ${THREADS}"
log "Workdir: ${WORKDIR}"
log "Logfile: ${LOGFILE}"
log "Mode: ${MODE}"
if [[ "${MODE}" == "PE" ]]; then
log "R1: ${R1}"
log "R2: ${R2}"
else
log "Reads: ${READS}"
fi
PM="$(pick_pm)"
log "Pkg manager: ${PM}"
ensure_env_exists
activate_env
log "Active envs:"
conda info --envs
log "Versions (if available):"
( python --version || true )
( checkm --version || true )
( gunc -v || true )
( minimap2 --version 2>&1 | head -n 2 || true )
( samtools --version 2>&1 | head -n 2 || true )
( mosdepth --version 2>&1 | head -n 2 || true )
( quast --version 2>&1 | head -n 2 || true )
( quast.py --version 2>&1 | head -n 2 || true )
# =========================
# Check/install missing tools in this env
MISSING_PKGS=()
for tool in minimap2 samtools mosdepth checkm gunc python; do
if ! need_cmd "${tool}"; then
MISSING_PKGS+=("${TOOL2PKG[$tool]}")
fi
done
QUAST_CMD="$(pick_quast_cmd)"
if [[ -z "${QUAST_CMD}" ]]; then
MISSING_PKGS+=("${TOOL2PKG[quast]}")
fi
if [[ "${#MISSING_PKGS[@]}" -gt 0 ]]; then
if [[ "${AUTO_INSTALL}" != "1" ]]; then
log "ERROR: missing tools and AUTO_INSTALL=0. Missing packages: ${MISSING_PKGS[*]}"
exit 1
fi
mapfile -t UNIQUE < <(printf "%s\n" "${MISSING_PKGS[@]}" | awk '!seen[$0]++')
install_pkgs_in_env "${PM}" "${UNIQUE[@]}"
activate_env
QUAST_CMD="$(pick_quast_cmd)"
fi
for tool in minimap2 samtools mosdepth checkm gunc python; do
need_cmd "${tool}" || { log "ERROR: still missing tool: ${tool}"; exit 1; }
done
[[ -n "${QUAST_CMD}" ]] || { log "ERROR: QUAST still missing."; exit 1; }
log "All tools ready. QUAST cmd: ${QUAST_CMD}"
# =========================
# Prepare workdir
mkdir -p "${WORKDIR}"/{genomes,reads,stats,quast,map,checkm,gunc,tmp}
ASM_ABS="$(realpath "${ASM}")"
ln -sf "${ASM_ABS}" "${WORKDIR}/genomes/${SAMPLE}.fasta"
if [[ "${MODE}" == "PE" ]]; then
R1_ABS="$(realpath "${R1}")"
R2_ABS="$(realpath "${R2}")"
ln -sf "${R1_ABS}" "${WORKDIR}/reads/${SAMPLE}.R1.fastq.gz"
ln -sf "${R2_ABS}" "${WORKDIR}/reads/${SAMPLE}.R2.fastq.gz"
else
READS_ABS="$(realpath "${READS}")"
ln -sf "${READS_ABS}" "${WORKDIR}/reads/${SAMPLE}.reads.fastq.gz"
fi
# =========================
# 1) QUAST
log "Run QUAST..."
"${QUAST_CMD}" "${WORKDIR}/genomes/${SAMPLE}.fasta" -o "${WORKDIR}/quast"
QUAST_TSV="${WORKDIR}/quast/report.tsv"
test -s "${QUAST_TSV}"
# =========================
# 2) Map reads + mosdepth
log "Map reads (minimap2) + sort BAM..."
SORT_T="$((THREADS>16?16:THREADS))"
if [[ "${MODE}" == "PE" ]]; then
minimap2 -t "${THREADS}" -ax sr \
"${WORKDIR}/genomes/${SAMPLE}.fasta" \
"${WORKDIR}/reads/${SAMPLE}.R1.fastq.gz" "${WORKDIR}/reads/${SAMPLE}.R2.fastq.gz" \
| samtools sort -@ "${SORT_T}" -o "${WORKDIR}/map/${SAMPLE}.bam" -
else
# legacy single-read mode; keep map-ont as in original script
minimap2 -t "${THREADS}" -ax map-ont \
"${WORKDIR}/genomes/${SAMPLE}.fasta" "${WORKDIR}/reads/${SAMPLE}.reads.fastq.gz" \
| samtools sort -@ "${SORT_T}" -o "${WORKDIR}/map/${SAMPLE}.bam" -
fi
samtools index "${WORKDIR}/map/${SAMPLE}.bam"
log "Compute depth (mosdepth)..."
mosdepth -t "${SORT_T}" "${WORKDIR}/map/${SAMPLE}" "${WORKDIR}/map/${SAMPLE}.bam"
MOS_SUMMARY="${WORKDIR}/map/${SAMPLE}.mosdepth.summary.txt"
test -s "${MOS_SUMMARY}"
# =========================
# 3) CheckM
log "Run CheckM lineage_wf..."
checkm lineage_wf -x fasta -t "${THREADS}" "${WORKDIR}/genomes" "${WORKDIR}/checkm/out"
log "Run CheckM qa..."
checkm qa "${WORKDIR}/checkm/out/lineage.ms" "${WORKDIR}/checkm/out" --tab_table -o 2 \
> "${WORKDIR}/checkm/checkm_summary.tsv"
CHECKM_SUM="${WORKDIR}/checkm/checkm_summary.tsv"
test -s "${CHECKM_SUM}"
# =========================
# 4) GUNC
log "Run GUNC..."
mkdir -p "${WORKDIR}/gunc/db" "${WORKDIR}/gunc/out"
if [[ -z "$(ls -A "${WORKDIR}/gunc/db" 2>/dev/null || true)" ]]; then
log "Downloading GUNC DB kind=${GUNC_DB_KIND} to ${WORKDIR}/gunc/db ..."
gunc download_db -db "${GUNC_DB_KIND}" "${WORKDIR}/gunc/db"
fi
DMND="$(find "${WORKDIR}/gunc/db" -type f -name "*.dmnd" | head -n 1 || true)"
if [[ -z "${DMND}" ]]; then
log "ERROR: No *.dmnd found under ${WORKDIR}/gunc/db after download."
ls -lah "${WORKDIR}/gunc/db" || true
exit 1
fi
log "Using GUNC db_file: ${DMND}"
gunc run \
--db_file "${DMND}" \
--input_fasta "${WORKDIR}/genomes/${SAMPLE}.fasta" \
--out_dir "${WORKDIR}/gunc/out" \
--threads "${THREADS}" \
--detailed_output \
--contig_taxonomy_output \
--use_species_level
ALL_LEVELS="$(find "${WORKDIR}/gunc/out" -name "*all_levels.tsv" | head -n 1 || true)"
test -n "${ALL_LEVELS}"
log "Found GUNC all_levels.tsv: ${ALL_LEVELS}"
# =========================
# 5) Parse outputs and write Table 1 TSV
log "Parse outputs → ${OUT_TSV}"
export SAMPLE WORKDIR OUT_TSV GUNC_ALL_LEVELS="${ALL_LEVELS}"
python - <<'PY'
import csv, os
sample = os.environ["SAMPLE"]
workdir = os.environ["WORKDIR"]
out_tsv = os.environ["OUT_TSV"]
gunc_all_levels = os.environ["GUNC_ALL_LEVELS"]
quast_tsv = os.path.join(workdir, "quast", "report.tsv")
mos_summary = os.path.join(workdir, "map", f"{sample}.mosdepth.summary.txt")
checkm_sum = os.path.join(workdir, "checkm", "checkm_summary.tsv")
def read_quast(path):
with open(path, newline="") as f:
rows = list(csv.reader(f, delimiter="\t"))
asm_idx = 1
d = {}
for r in rows[1:]:
if not r: continue
key = r[0].strip()
val = r[asm_idx].strip() if asm_idx < len(r) else ""
d[key] = val
return d
def read_mosdepth(path):
with open(path) as f:
for line in f:
if line.startswith("chrom"): continue
parts = line.rstrip("\n").split("\t")
if len(parts) >= 4 and parts[0] == "total":
return parts[3]
return ""
def read_checkm(path, sample):
with open(path, newline="") as f:
reader = csv.DictReader(f, delimiter="\t")
for row in reader:
bid = row.get("Bin Id") or row.get("Bin") or row.get("bin_id") or ""
if bid == sample:
return row
return {}
def read_gunc_all_levels(path):
coarse_lvls = {"kingdom","phylum","class"}
fine_lvls = {"order","family","genus","species"}
coarse, fine = [], []
best_line = None
rank = {"kingdom":0,"phylum":1,"class":2,"order":3,"family":4,"genus":5,"species":6}
best_rank = -1
with open(path, newline="") as f:
reader = csv.DictReader(f, delimiter="\t")
for row in reader:
lvl = (row.get("taxonomic_level") or "").strip()
p = row.get("proportion_genes_retained_in_major_clades") or ""
try:
pv = float(p)
except:
pv = None
if pv is not None:
if lvl in coarse_lvls: coarse.append(pv)
if lvl in fine_lvls: fine.append(pv)
if lvl in rank and rank[lvl] > best_rank:
best_rank = rank[lvl]
best_line = row
coarse_mean = sum(coarse)/len(coarse) if coarse else ""
fine_mean = sum(fine)/len(fine) if fine else ""
contamination_portion = best_line.get("contamination_portion","") if best_line else ""
pass_gunc = best_line.get("pass.GUNC","") if best_line else ""
return coarse_mean, fine_mean, contamination_portion, pass_gunc
qu = read_quast(quast_tsv)
mean_depth = read_mosdepth(mos_summary)
ck = read_checkm(checkm_sum, sample)
coarse_mean, fine_mean, contamination_portion, pass_gunc = read_gunc_all_levels(gunc_all_levels)
header = [
"Sample",
"Genome_length_bp",
"Contigs",
"N50_bp",
"L50",
"GC_percent",
"Mean_depth_x",
"CheckM_completeness_percent",
"CheckM_contamination_percent",
"CheckM_strain_heterogeneity_percent",
"GUNC_coarse_consistency",
"GUNC_fine_consistency",
"GUNC_contamination_portion",
"GUNC_pass"
]
row = [
sample,
qu.get("Total length", ""),
qu.get("# contigs", ""),
qu.get("N50", ""),
qu.get("L50", ""),
qu.get("GC (%)", ""),
mean_depth,
ck.get("Completeness", ""),
ck.get("Contamination", ""),
ck.get("Strain heterogeneity", ""),
f"{coarse_mean:.4f}" if isinstance(coarse_mean, float) else coarse_mean,
f"{fine_mean:.4f}" if isinstance(fine_mean, float) else fine_mean,
contamination_portion,
pass_gunc
]
with open(out_tsv, "w", newline="") as f:
w = csv.writer(f, delimiter="\t")
w.writerow(header)
w.writerow(row)
print(f"OK: wrote {out_tsv}")
PY
log "SUCCESS"
log "Output TSV: ${OUT_TSV}"
log "Workdir: ${WORKDIR}"
log "Logfile: ${LOGFILE}"export_table1_stats_to_excel_py36_compat.py
#!/usr/bin/env python3
# -*- coding: utf-8 -*-
"""
Export a comprehensive Excel workbook from a Table1 pipeline workdir.
Python 3.6 compatible (no PEP604 unions, no builtin generics).
Requires: openpyxl
Sheets (as available):
- Summary
- Table1 (if Table1_*.tsv exists)
- QUAST_report (report.tsv)
- QUAST_metrics (metric/value)
- Mosdepth_summary (*.mosdepth.summary.txt)
- CheckM (checkm_summary.tsv)
- GUNC_* (all .tsv under gunc/out)
- File_Inventory (relative path, size, mtime; optional md5 for small files)
- Run_log_preview (head/tail of latest log under workdir/logs or workdir/*/logs)
"""
from __future__ import print_function
import argparse
import csv
import hashlib
import os
import sys
import time
from pathlib import Path
try:
from openpyxl import Workbook
from openpyxl.utils import get_column_letter
except ImportError:
sys.stderr.write("ERROR: openpyxl is required. Install with:\n"
" conda install -c conda-forge openpyxl\n")
raise
MAX_XLSX_ROWS = 1048576
def safe_sheet_name(name, used):
# Excel: <=31 chars, cannot contain: : \ / ? * [ ]
bad = r'[:\\/?*\[\]]'
base = name.strip() or "Sheet"
base = __import__("re").sub(bad, "_", base)
base = base[:31]
if base not in used:
used.add(base)
return base
# make unique with suffix
for i in range(2, 1000):
suffix = "_%d" % i
cut = 31 - len(suffix)
candidate = (base[:cut] + suffix)
if candidate not in used:
used.add(candidate)
return candidate
raise RuntimeError("Too many duplicate sheet names for base=%s" % base)
def autosize(ws, max_width=60):
for col in ws.columns:
max_len = 0
col_letter = get_column_letter(col[0].column)
for cell in col:
v = cell.value
if v is None:
continue
s = str(v)
if len(s) > max_len:
max_len = len(s)
ws.column_dimensions[col_letter].width = min(max_width, max(10, max_len + 2))
def write_table(ws, header, rows, max_rows=None):
if header:
ws.append(header)
count = 0
for r in rows:
ws.append(r)
count += 1
if max_rows is not None and count >= max_rows:
break
def read_tsv(path, max_rows=None):
header = []
rows = []
with path.open("r", newline="") as f:
reader = csv.reader(f, delimiter="\t")
for i, r in enumerate(reader):
if i == 0:
header = r
continue
rows.append(r)
if max_rows is not None and len(rows) >= max_rows:
break
return header, rows
def read_text_table(path, max_rows=None):
# for mosdepth summary (tsv with header)
return read_tsv(path, max_rows=max_rows)
def md5_file(path, chunk=1024*1024):
h = hashlib.md5()
with path.open("rb") as f:
while True:
b = f.read(chunk)
if not b:
break
h.update(b)
return h.hexdigest()
def find_latest_log(workdir):
candidates = []
# common locations
for p in [workdir / "logs", workdir / "log", workdir / "Logs"]:
if p.exists():
candidates.extend(p.glob("*.log"))
# nested logs
candidates.extend(workdir.glob("**/logs/*.log"))
if not candidates:
return None
candidates.sort(key=lambda x: x.stat().st_mtime, reverse=True)
return candidates[0]
def add_summary_sheet(wb, used, info_items):
ws = wb.create_sheet(title=safe_sheet_name("Summary", used))
ws.append(["Key", "Value"])
for k, v in info_items:
ws.append([k, v])
autosize(ws)
def add_log_preview(wb, used, log_path, head_n=80, tail_n=120):
if log_path is None or not log_path.exists():
return
ws = wb.create_sheet(title=safe_sheet_name("Run_log_preview", used))
ws.append(["Log path", str(log_path)])
ws.append([])
lines = log_path.read_text(errors="replace").splitlines()
ws.append(["--- HEAD (%d) ---" % head_n])
for line in lines[:head_n]:
ws.append([line])
ws.append([])
ws.append(["--- TAIL (%d) ---" % tail_n])
for line in lines[-tail_n:]:
ws.append([line])
ws.column_dimensions["A"].width = 120
def add_file_inventory(wb, used, workdir, do_md5=True, md5_max_bytes=200*1024*1024, max_rows=None):
ws = wb.create_sheet(title=safe_sheet_name("File_Inventory", used))
ws.append(["relative_path", "size_bytes", "mtime_iso", "md5(optional)"])
count = 0
for p in sorted(workdir.rglob("*")):
if p.is_dir():
continue
rel = str(p.relative_to(workdir))
st = p.stat()
mtime = time.strftime("%Y-%m-%d %H:%M:%S", time.localtime(st.st_mtime))
md5 = ""
if do_md5 and st.st_size <= md5_max_bytes:
try:
md5 = md5_file(p)
except Exception:
md5 = "ERROR"
ws.append([rel, st.st_size, mtime, md5])
count += 1
if max_rows is not None and count >= max_rows:
break
autosize(ws, max_width=80)
def add_tsv_sheet(wb, used, name, path, max_rows=None):
header, rows = read_tsv(path, max_rows=max_rows)
ws = wb.create_sheet(title=safe_sheet_name(name, used))
write_table(ws, header, rows, max_rows=max_rows)
autosize(ws, max_width=80)
def add_quast_metrics_sheet(wb, used, quast_report_tsv):
header, rows = read_tsv(quast_report_tsv, max_rows=None)
if not header or len(header) < 2:
return
asm_name = header[1]
ws = wb.create_sheet(title=safe_sheet_name("QUAST_metrics", used))
ws.append(["Metric", asm_name])
for r in rows:
if not r:
continue
metric = r[0]
val = r[1] if len(r) > 1 else ""
ws.append([metric, val])
autosize(ws, max_width=80)
def main():
ap = argparse.ArgumentParser()
ap.add_argument("--workdir", required=True, help="workdir produced by pipeline (e.g., table1_GE11174_work)")
ap.add_argument("--out", required=True, help="output .xlsx")
ap.add_argument("--sample", default="", help="sample name for summary")
ap.add_argument("--max-rows", type=int, default=200000, help="max rows per large sheet")
ap.add_argument("--no-md5", action="store_true", help="skip md5 calculation in File_Inventory")
args = ap.parse_args()
workdir = Path(args.workdir).resolve()
out = Path(args.out).resolve()
if not workdir.exists():
sys.stderr.write("ERROR: workdir not found: %s\n" % workdir)
sys.exit(2)
wb = Workbook()
# remove default sheet
wb.remove(wb.active)
used = set()
# Summary info
info = [
("sample", args.sample or ""),
("workdir", str(workdir)),
("generated_at", time.strftime("%Y-%m-%d %H:%M:%S")),
("python", sys.version.replace("\n", " ")),
("openpyxl", __import__("openpyxl").__version__),
]
add_summary_sheet(wb, used, info)
# Table1 TSV (try common names)
table1_candidates = list(workdir.glob("Table1_*.tsv")) + list(workdir.glob("*.tsv"))
# Prefer Table1_*.tsv in workdir root
table1_path = None
for p in table1_candidates:
if p.name.startswith("Table1_") and p.suffix == ".tsv":
table1_path = p
break
if table1_path is None:
# maybe created in cwd, not inside workdir; try alongside workdir
parent = workdir.parent
for p in parent.glob("Table1_*.tsv"):
if args.sample and args.sample in p.name:
table1_path = p
break
if table1_path is None and list(parent.glob("Table1_*.tsv")):
table1_path = sorted(parent.glob("Table1_*.tsv"))[0]
if table1_path is not None and table1_path.exists():
add_tsv_sheet(wb, used, "Table1", table1_path, max_rows=args.max_rows)
# QUAST
quast_report = workdir / "quast" / "report.tsv"
if quast_report.exists():
add_tsv_sheet(wb, used, "QUAST_report", quast_report, max_rows=args.max_rows)
add_quast_metrics_sheet(wb, used, quast_report)
# Mosdepth summary
for p in sorted((workdir / "map").glob("*.mosdepth.summary.txt")):
# mosdepth summary is TSV-like
name = "Mosdepth_" + p.stem.replace(".mosdepth.summary", "")
add_tsv_sheet(wb, used, name[:31], p, max_rows=args.max_rows)
# CheckM
checkm_sum = workdir / "checkm" / "checkm_summary.tsv"
if checkm_sum.exists():
add_tsv_sheet(wb, used, "CheckM", checkm_sum, max_rows=args.max_rows)
# GUNC outputs (all TSV under gunc/out)
gunc_out = workdir / "gunc" / "out"
if gunc_out.exists():
for p in sorted(gunc_out.rglob("*.tsv")):
rel = str(p.relative_to(gunc_out))
sheet = "GUNC_" + rel.replace("/", "_").replace("\\", "_").replace(".tsv", "")
add_tsv_sheet(wb, used, sheet[:31], p, max_rows=args.max_rows)
# Log preview
latest_log = find_latest_log(workdir)
add_log_preview(wb, used, latest_log)
# File inventory
add_file_inventory(
wb, used, workdir,
do_md5=(not args.no_md5),
md5_max_bytes=200*1024*1024,
max_rows=args.max_rows
)
# Save
out.parent.mkdir(parents=True, exist_ok=True)
wb.save(str(out))
print("OK: wrote %s" % out)
if __name__ == "__main__":
main()run_resistome_virulome_dedup.sh
#!/usr/bin/env bash
set -Eeuo pipefail
# -------- user inputs --------
ENV_NAME="${ENV_NAME:-bengal3_ac3}"
ASM="${ASM:-GE11174.fasta}"
SAMPLE="${SAMPLE:-GE11174}"
OUTDIR="${OUTDIR:-resistome_virulence_${SAMPLE}}"
THREADS="${THREADS:-16}"
# thresholds (set to 0/0 if you truly want ABRicate defaults)
MINID="${MINID:-90}"
MINCOV="${MINCOV:-60}"
# ----------------------------
log(){ echo "[$(date +'%F %T')] $*" >&2; }
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
activate_env() {
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
conda activate "${ENV_NAME}"
}
main(){
activate_env
mkdir -p "${OUTDIR}"/{raw,amr,virulence,card,tmp}
log "Env: ${ENV_NAME}"
log "ASM: ${ASM}"
log "Sample: ${SAMPLE}"
log "Outdir: ${OUTDIR}"
log "ABRicate thresholds: MINID=${MINID} MINCOV=${MINCOV}"
log "ABRicate DB list:"
abricate --list | egrep -i "vfdb|resfinder|megares|card" || true
# Make sure indices exist
log "Running abricate --setupdb (safe even if already done)..."
abricate --setupdb
# ---- ABRicate AMR DBs ----
log "Running ABRicate: ResFinder"
abricate --db resfinder --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
log "Running ABRicate: MEGARes"
abricate --db megares --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.megares.tab"
# ---- Virulence (VFDB) ----
log "Running ABRicate: VFDB"
abricate --db vfdb --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
# ---- CARD: prefer RGI if available, else ABRicate card ----
CARD_MODE="ABRicate"
if need_cmd rgi; then
log "RGI found. Trying RGI (CARD) ..."
set +e
rgi main --input_sequence "${ASM}" --output_file "${OUTDIR}/card/${SAMPLE}.rgi" --input_type contig --num_threads "${THREADS}"
rc=$?
set -e
if [[ $rc -eq 0 ]]; then
CARD_MODE="RGI"
else
log "RGI failed (likely CARD data not installed). Falling back to ABRicate card."
fi
fi
if [[ "${CARD_MODE}" == "ABRicate" ]]; then
log "Running ABRicate: CARD"
abricate --db card --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.card.tab"
fi
# ---- Build deduplicated tables ----
log "Creating deduplicated AMR/VFDB tables..."
export OUTDIR SAMPLE CARD_MODE
python - <<'PY'
import os, re
from pathlib import Path
import pandas as pd
from io import StringIO
outdir = Path(os.environ["OUTDIR"])
sample = os.environ["SAMPLE"]
card_mode = os.environ["CARD_MODE"]
def read_abricate_tab(path: Path, source: str) -> pd.DataFrame:
if not path.exists() or path.stat().st_size == 0:
return pd.DataFrame()
lines=[]
with path.open("r", errors="replace") as f:
for line in f:
if line.startswith("#") or not line.strip():
continue
lines.append(line)
if not lines:
return pd.DataFrame()
df = pd.read_csv(StringIO("".join(lines)), sep="\t", dtype=str)
df.insert(0, "Source", source)
return df
def to_num(s):
try:
return float(str(s).replace("%",""))
except:
return None
def normalize_abricate(df: pd.DataFrame, dbname: str) -> pd.DataFrame:
if df.empty:
return pd.DataFrame(columns=[
"Source","Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"
])
# Column names vary slightly; handle common ones
gene = "GENE" if "GENE" in df.columns else None
prod = "PRODUCT" if "PRODUCT" in df.columns else None
acc = "ACCESSION" if "ACCESSION" in df.columns else None
contig = "SEQUENCE" if "SEQUENCE" in df.columns else ("CONTIG" if "CONTIG" in df.columns else None)
start = "START" if "START" in df.columns else None
end = "END" if "END" in df.columns else None
strand= "STRAND" if "STRAND" in df.columns else None
pid = "%IDENTITY" if "%IDENTITY" in df.columns else ("% Identity" if "% Identity" in df.columns else None)
pcv = "%COVERAGE" if "%COVERAGE" in df.columns else ("% Coverage" if "% Coverage" in df.columns else None)
out = pd.DataFrame()
out["Source"] = df["Source"]
out["Database"] = dbname
out["Gene"] = df[gene] if gene else ""
out["Product"] = df[prod] if prod else ""
out["Accession"] = df[acc] if acc else ""
out["Contig"] = df[contig] if contig else ""
out["Start"] = df[start] if start else ""
out["End"] = df[end] if end else ""
out["Strand"] = df[strand] if strand else ""
out["Pct_Identity"] = df[pid] if pid else ""
out["Pct_Coverage"] = df[pcv] if pcv else ""
return out
def dedup_best(df: pd.DataFrame, key_cols):
"""Keep best hit per key by highest identity, then coverage, then longest span."""
if df.empty:
return df
# numeric helpers
df = df.copy()
df["_pid"] = df["Pct_Identity"].map(to_num)
df["_pcv"] = df["Pct_Coverage"].map(to_num)
def span(row):
try:
return abs(int(row["End"]) - int(row["Start"])) + 1
except:
return 0
df["_span"] = df.apply(span, axis=1)
# sort best-first
df = df.sort_values(by=["_pid","_pcv","_span"], ascending=[False,False,False], na_position="last")
df = df.drop_duplicates(subset=key_cols, keep="first")
df = df.drop(columns=["_pid","_pcv","_span"])
return df
# ---------- AMR inputs ----------
amr_frames = []
# ResFinder (often 0 hits; still okay)
resfinder = outdir / "raw" / f"{sample}.resfinder.tab"
df = read_abricate_tab(resfinder, "ABRicate")
amr_frames.append(normalize_abricate(df, "ResFinder"))
# MEGARes
megares = outdir / "raw" / f"{sample}.megares.tab"
df = read_abricate_tab(megares, "ABRicate")
amr_frames.append(normalize_abricate(df, "MEGARes"))
# CARD: RGI or ABRicate
if card_mode == "RGI":
# Try common RGI tab outputs
prefix = outdir / "card" / f"{sample}.rgi"
rgi_tab = None
for ext in [".txt",".tab",".tsv"]:
p = Path(str(prefix) + ext)
if p.exists() and p.stat().st_size > 0:
rgi_tab = p
break
if rgi_tab is not None:
rgi = pd.read_csv(rgi_tab, sep="\t", dtype=str)
out = pd.DataFrame()
out["Source"] = "RGI"
out["Database"] = "CARD"
# Prefer ARO_name/Best_Hit_ARO if present
out["Gene"] = rgi["ARO_name"] if "ARO_name" in rgi.columns else (rgi["Best_Hit_ARO"] if "Best_Hit_ARO" in rgi.columns else "")
out["Product"] = rgi["ARO_name"] if "ARO_name" in rgi.columns else ""
out["Accession"] = rgi["ARO_accession"] if "ARO_accession" in rgi.columns else ""
out["Contig"] = rgi["Sequence"] if "Sequence" in rgi.columns else ""
out["Start"] = rgi["Start"] if "Start" in rgi.columns else ""
out["End"] = rgi["Stop"] if "Stop" in rgi.columns else (rgi["End"] if "End" in rgi.columns else "")
out["Strand"] = rgi["Orientation"] if "Orientation" in rgi.columns else ""
out["Pct_Identity"] = rgi["% Identity"] if "% Identity" in rgi.columns else ""
out["Pct_Coverage"] = rgi["% Coverage"] if "% Coverage" in rgi.columns else ""
amr_frames.append(out)
else:
card = outdir / "raw" / f"{sample}.card.tab"
df = read_abricate_tab(card, "ABRicate")
amr_frames.append(normalize_abricate(df, "CARD"))
amr_all = pd.concat([x for x in amr_frames if not x.empty], ignore_index=True) if any(not x.empty for x in amr_frames) else pd.DataFrame(
columns=["Source","Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"]
)
# Deduplicate within each (Database,Gene) – this is usually what you want for manuscript tables
amr_dedup = dedup_best(amr_all, key_cols=["Database","Gene"])
# Sort nicely
if not amr_dedup.empty:
amr_dedup = amr_dedup.sort_values(["Database","Gene"]).reset_index(drop=True)
amr_out = outdir / "Table_AMR_genes_dedup.tsv"
amr_dedup.to_csv(amr_out, sep="\t", index=False)
# ---------- Virulence (VFDB) ----------
vfdb = outdir / "raw" / f"{sample}.vfdb.tab"
vf = read_abricate_tab(vfdb, "ABRicate")
vf_norm = normalize_abricate(vf, "VFDB")
# Dedup within (Gene) for VFDB (or use Database,Gene; Database constant)
vf_dedup = dedup_best(vf_norm, key_cols=["Gene"]) if not vf_norm.empty else vf_norm
if not vf_dedup.empty:
vf_dedup = vf_dedup.sort_values(["Gene"]).reset_index(drop=True)
vf_out = outdir / "Table_Virulence_VFDB_dedup.tsv"
vf_dedup.to_csv(vf_out, sep="\t", index=False)
print("OK wrote:")
print(" ", amr_out)
print(" ", vf_out)
PY
log "Done."
log "Outputs:"
log " ${OUTDIR}/Table_AMR_genes_dedup.tsv"
log " ${OUTDIR}/Table_Virulence_VFDB_dedup.tsv"
log "Raw:"
log " ${OUTDIR}/raw/${SAMPLE}.*.tab"
}
mainrun_abricate_resistome_virulome_one_per_gene.sh
#!/usr/bin/env bash
set -Eeuo pipefail
# ------------------- USER SETTINGS -------------------
ENV_NAME="${ENV_NAME:-bengal3_ac3}"
ASM="${ASM:-GE11174.fasta}" # input assembly fasta
SAMPLE="${SAMPLE:-GE11174}"
OUTDIR="${OUTDIR:-resistome_virulence_${SAMPLE}}"
THREADS="${THREADS:-16}"
# ABRicate thresholds
# If you want your earlier "35 genes" behavior, use MINID=70 MINCOV=50.
# If you want stricter: e.g. MINID=80 MINCOV=70.
MINID="${MINID:-70}"
MINCOV="${MINCOV:-50}"
# -----------------------------------------------------
ts(){ date +"%F %T"; }
log(){ echo "[$(ts)] $*" >&2; }
on_err(){
local ec=$?
log "ERROR: failed (exit=${ec}) at line ${BASH_LINENO[0]}: ${BASH_COMMAND}"
exit $ec
}
trap on_err ERR
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
activate_env() {
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
conda activate "${ENV_NAME}"
}
main(){
activate_env
log "Env: ${ENV_NAME}"
log "ASM: ${ASM}"
log "Sample: ${SAMPLE}"
log "Outdir: ${OUTDIR}"
log "Threads: ${THREADS}"
log "ABRicate thresholds: MINID=${MINID} MINCOV=${MINCOV}"
mkdir -p "${OUTDIR}"/{raw,logs}
# Save full log
LOGFILE="${OUTDIR}/logs/run_$(date +'%F_%H%M%S').log"
exec > >(tee -a "${LOGFILE}") 2>&1
log "Tool versions:"
abricate --version || true
abricate-get_db --help | head -n 5 || true
log "ABRicate DB list (selected):"
abricate --list | egrep -i "vfdb|resfinder|megares|card" || true
log "Indexing ABRicate databases (safe to re-run)..."
abricate --setupdb
# ---------------- Run ABRicate ----------------
log "Running ABRicate: MEGARes"
abricate --db megares --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.megares.tab"
log "Running ABRicate: CARD"
abricate --db card --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.card.tab"
log "Running ABRicate: ResFinder"
abricate --db resfinder --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
log "Running ABRicate: VFDB"
abricate --db vfdb --minid "${MINID}" --mincov "${MINCOV}" "${ASM}" > "${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
# --------------- Build tables -----------------
export OUTDIR SAMPLE
export MEGARES_TAB="${OUTDIR}/raw/${SAMPLE}.megares.tab"
export CARD_TAB="${OUTDIR}/raw/${SAMPLE}.card.tab"
export RESFINDER_TAB="${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
export VFDB_TAB="${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
export AMR_OUT="${OUTDIR}/Table_AMR_genes_one_per_gene.tsv"
export VIR_OUT="${OUTDIR}/Table_Virulence_VFDB_dedup.tsv"
export STATUS_OUT="${OUTDIR}/Table_DB_hit_counts.tsv"
log "Generating deduplicated tables..."
python - <<'PY'
import os
import pandas as pd
from pathlib import Path
megares_tab = Path(os.environ["MEGARES_TAB"])
card_tab = Path(os.environ["CARD_TAB"])
resfinder_tab = Path(os.environ["RESFINDER_TAB"])
vfdb_tab = Path(os.environ["VFDB_TAB"])
amr_out = Path(os.environ["AMR_OUT"])
vir_out = Path(os.environ["VIR_OUT"])
status_out = Path(os.environ["STATUS_OUT"])
def read_abricate(path: Path) -> pd.DataFrame:
"""Parse ABRicate .tab where header line starts with '#FILE'."""
if (not path.exists()) or path.stat().st_size == 0:
return pd.DataFrame()
header = None
rows = []
with path.open("r", errors="replace") as f:
for line in f:
if not line.strip():
continue
if line.startswith("#FILE"):
header = line.lstrip("#").rstrip("\n").split("\t")
continue
if line.startswith("#"):
continue
rows.append(line.rstrip("\n").split("\t"))
if header is None:
return pd.DataFrame()
if not rows:
return pd.DataFrame(columns=header)
return pd.DataFrame(rows, columns=header)
def normalize(df: pd.DataFrame, dbname: str) -> pd.DataFrame:
cols_out = ["Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"]
if df is None or df.empty:
return pd.DataFrame(columns=cols_out)
out = pd.DataFrame({
"Database": dbname,
"Gene": df.get("GENE",""),
"Product": df.get("PRODUCT",""),
"Accession": df.get("ACCESSION",""),
"Contig": df.get("SEQUENCE",""),
"Start": df.get("START",""),
"End": df.get("END",""),
"Strand": df.get("STRAND",""),
"Pct_Identity": pd.to_numeric(df.get("%IDENTITY",""), errors="coerce"),
"Pct_Coverage": pd.to_numeric(df.get("%COVERAGE",""), errors="coerce"),
})
return out[cols_out]
def best_hit_dedup(df: pd.DataFrame, key_cols):
"""Keep best hit by highest identity, then coverage, then alignment length."""
if df.empty:
return df
d = df.copy()
d["Start_i"] = pd.to_numeric(d["Start"], errors="coerce").fillna(0).astype(int)
d["End_i"] = pd.to_numeric(d["End"], errors="coerce").fillna(0).astype(int)
d["Len"] = (d["End_i"] - d["Start_i"]).abs() + 1
d = d.sort_values(["Pct_Identity","Pct_Coverage","Len"], ascending=[False,False,False])
d = d.drop_duplicates(subset=key_cols, keep="first")
return d.drop(columns=["Start_i","End_i","Len"])
def count_hits(path: Path) -> int:
if not path.exists():
return 0
n = 0
with path.open() as f:
for line in f:
if line.startswith("#") or not line.strip():
continue
n += 1
return n
# -------- load + normalize --------
parts = []
for dbname, p in [("MEGARes", megares_tab), ("CARD", card_tab), ("ResFinder", resfinder_tab)]:
df = read_abricate(p)
parts.append(normalize(df, dbname))
amr_all = pd.concat([x for x in parts if not x.empty], ignore_index=True) if any(not x.empty for x in parts) else pd.DataFrame(
columns=["Database","Gene","Product","Accession","Contig","Start","End","Strand","Pct_Identity","Pct_Coverage"]
)
# remove empty genes
amr_all = amr_all[amr_all["Gene"].astype(str).str.len() > 0].copy()
# best per (Database,Gene)
amr_db_gene = best_hit_dedup(amr_all, ["Database","Gene"]) if not amr_all.empty else amr_all
# one row per Gene overall, priority: CARD > ResFinder > MEGARes
priority = {"CARD": 0, "ResFinder": 1, "MEGARes": 2}
if not amr_db_gene.empty:
amr_db_gene["prio"] = amr_db_gene["Database"].map(priority).fillna(9).astype(int)
amr_one = amr_db_gene.sort_values(
["Gene","prio","Pct_Identity","Pct_Coverage"],
ascending=[True, True, False, False]
)
amr_one = amr_one.drop_duplicates(["Gene"], keep="first").drop(columns=["prio"])
amr_one = amr_one.sort_values(["Gene"]).reset_index(drop=True)
else:
amr_one = amr_db_gene
amr_out.parent.mkdir(parents=True, exist_ok=True)
amr_one.to_csv(amr_out, sep="\t", index=False)
# -------- VFDB --------
vf = normalize(read_abricate(vfdb_tab), "VFDB")
vf = vf[vf["Gene"].astype(str).str.len() > 0].copy()
vf_one = best_hit_dedup(vf, ["Gene"]) if not vf.empty else vf
if not vf_one.empty:
vf_one = vf_one.sort_values(["Gene"]).reset_index(drop=True)
vir_out.parent.mkdir(parents=True, exist_ok=True)
vf_one.to_csv(vir_out, sep="\t", index=False)
# -------- status counts --------
status = pd.DataFrame([
{"Database":"MEGARes", "Hit_lines": count_hits(megares_tab), "File": str(megares_tab)},
{"Database":"CARD", "Hit_lines": count_hits(card_tab), "File": str(card_tab)},
{"Database":"ResFinder", "Hit_lines": count_hits(resfinder_tab), "File": str(resfinder_tab)},
{"Database":"VFDB", "Hit_lines": count_hits(vfdb_tab), "File": str(vfdb_tab)},
])
status_out.parent.mkdir(parents=True, exist_ok=True)
status.to_csv(status_out, sep="\t", index=False)
print("OK wrote:")
print(" ", amr_out, "rows=", len(amr_one))
print(" ", vir_out, "rows=", len(vf_one))
print(" ", status_out)
PY
log "Finished."
log "Main outputs:"
log " ${AMR_OUT}"
log " ${VIR_OUT}"
log " ${STATUS_OUT}"
log "Raw ABRicate outputs:"
log " ${OUTDIR}/raw/${SAMPLE}.megares.tab"
log " ${OUTDIR}/raw/${SAMPLE}.card.tab"
log " ${OUTDIR}/raw/${SAMPLE}.resfinder.tab"
log " ${OUTDIR}/raw/${SAMPLE}.vfdb.tab"
log "Log:"
log " ${LOGFILE}"
}
mainresolve_best_assemblies_entrez.py
#!/usr/bin/env python3
import csv
import os
import re
import sys
import time
from dataclasses import dataclass
from typing import List, Optional, Tuple
from Bio import Entrez
# REQUIRED by NCBI policy
Entrez.email = os.environ.get("NCBI_EMAIL", "your.email@example.com")
# Be nice to NCBI
ENTREZ_DELAY_SEC = float(os.environ.get("ENTREZ_DELAY_SEC", "0.34"))
LEVEL_RANK = {
"Complete Genome": 0,
"Chromosome": 1,
"Scaffold": 2,
"Contig": 3,
# sometimes NCBI uses slightly different strings:
"complete genome": 0,
"chromosome": 1,
"scaffold": 2,
"contig": 3,
}
def level_rank(level: str) -> int:
return LEVEL_RANK.get(level.strip(), 99)
def is_refseq(accession: str) -> bool:
return accession.startswith("GCF_")
@dataclass
class AssemblyHit:
assembly_uid: str
assembly_accession: str # GCF_... or GCA_...
organism: str
strain: str
assembly_level: str
refseq_category: str
submitter: str
ftp_path: str
def entrez_search_assembly(term: str, retmax: int = 50) -> List[str]:
"""Return Assembly UIDs matching term."""
h = Entrez.esearch(db="assembly", term=term, retmax=str(retmax))
rec = Entrez.read(h)
h.close()
time.sleep(ENTREZ_DELAY_SEC)
return rec.get("IdList", [])
def entrez_esummary_assembly(uids: List[str]) -> List[AssemblyHit]:
"""Fetch assembly summary records for given UIDs."""
if not uids:
return []
h = Entrez.esummary(db="assembly", id=",".join(uids), report="full")
rec = Entrez.read(h)
h.close()
time.sleep(ENTREZ_DELAY_SEC)
hits: List[AssemblyHit] = []
docs = rec.get("DocumentSummarySet", {}).get("DocumentSummary", [])
for d in docs:
# Some fields can be missing
acc = str(d.get("AssemblyAccession", "")).strip()
org = str(d.get("Organism", "")).strip()
level = str(d.get("AssemblyStatus", "")).strip() or str(d.get("AssemblyLevel", "")).strip()
# NCBI uses "AssemblyStatus" sometimes, "AssemblyLevel" other times;
# in practice AssemblyStatus often equals "Complete Genome"/"Chromosome"/...
if not level:
level = str(d.get("AssemblyLevel", "")).strip()
strain = str(d.get("Biosample", "")).strip()
# Strain is not always in a clean field. Try "Sub_value" in Meta, or parse Submitter/Title.
# We'll try a few common places:
title = str(d.get("AssemblyName", "")).strip()
submitter = str(d.get("SubmitterOrganization", "")).strip()
refcat = str(d.get("RefSeq_category", "")).strip()
ftp = str(d.get("FtpPath_RefSeq", "")).strip() or str(d.get("FtpPath_GenBank", "")).strip()
hits.append(
AssemblyHit(
assembly_uid=str(d.get("Uid", "")),
assembly_accession=acc,
organism=org,
strain=strain,
assembly_level=level,
refseq_category=refcat,
submitter=submitter,
ftp_path=ftp,
)
)
return hits
def best_hit(hits: List[AssemblyHit]) -> Optional[AssemblyHit]:
"""Pick best hit by level (Complete>Chromosome>...), prefer RefSeq, then prefer representative/reference."""
if not hits:
return None
def key(h: AssemblyHit) -> Tuple[int, int, int, str]:
# lower is better
lvl = level_rank(h.assembly_level)
ref = 0 if is_refseq(h.assembly_accession) else 1
# prefer reference/representative if present
cat = (h.refseq_category or "").lower()
rep = 0
if "reference" in cat:
rep = 0
elif "representative" in cat:
rep = 1
else:
rep = 2
# tie-breaker: accession string (stable)
return (lvl, ref, rep, h.assembly_accession)
return sorted(hits, key=key)[0]
def relaxed_fallback_terms(organism: str, strain_tokens: List[str]) -> List[str]:
"""
Build fallback search terms:
1) organism + strain tokens
2) organism only (species-only)
3) genus-only (if species fails)
"""
terms = []
# 1) Full term: organism + strain tokens
if strain_tokens:
t = f'"{organism}"[Organism] AND (' + " OR ".join(f'"{s}"[All Fields]' for s in strain_tokens) + ")"
terms.append(t)
# 2) Species only
terms.append(f'"{organism}"[Organism]')
# 3) Genus only
genus = organism.split()[0]
terms.append(f'"{genus}"[Organism]')
return terms
def resolve_one(label: str, organism: str, strain_tokens: List[str], retmax: int = 80) -> Tuple[str, Optional[AssemblyHit], str]:
"""
Returns:
- selected accession or "NA"
- selected hit (optional)
- which query term matched
"""
for term in relaxed_fallback_terms(organism, strain_tokens):
uids = entrez_search_assembly(term, retmax=retmax)
hits = entrez_esummary_assembly(uids)
chosen = best_hit(hits)
if chosen and chosen.assembly_accession:
return chosen.assembly_accession, chosen, term
return "NA", None, ""
def parse_targets_tsv(path: str) -> List[Tuple[str, str, List[str]]]:
"""
Input TSV format:
label organism strain_tokens
where strain_tokens is a semicolon-separated list, e.g. "FRB97;FRB 97"
"""
rows = []
with open(path, newline="") as f:
r = csv.DictReader(f, delimiter="\t")
for row in r:
label = row["label"].strip()
org = row["organism"].strip()
tokens = [x.strip() for x in row.get("strain_tokens", "").split(";") if x.strip()]
rows.append((label, org, tokens))
return rows
def main():
if len(sys.argv) < 3:
print("Usage: resolve_best_assemblies_entrez.py targets.tsv out.tsv", file=sys.stderr)
sys.exit(2)
targets_tsv = sys.argv[1]
out_tsv = sys.argv[2]
targets = parse_targets_tsv(targets_tsv)
with open(out_tsv, "w", newline="") as f:
w = csv.writer(f, delimiter="\t")
w.writerow(["label", "best_accession", "assembly_level", "refseq_category", "organism", "query_used"])
for label, org, tokens in targets:
acc, hit, term = resolve_one(label, org, tokens)
if hit:
w.writerow([label, acc, hit.assembly_level, hit.refseq_category, hit.organism, term])
print(f"[OK] {label} -> {acc} ({hit.assembly_level})")
else:
w.writerow([label, "NA", "", "", org, ""])
print(f"[WARN] {label} -> NA (no assemblies found)")
if __name__ == "__main__":
main()build_wgs_tree_fig3B.sh
#!/usr/bin/env bash
set -euo pipefail
###############################################################################
# Core-genome phylogeny pipeline (genome-wide; no 16S/MLST):
#
# Uses existing conda env prefix:
# ENV_NAME=/home/jhuang/miniconda3/envs/bengal3_ac3
#
# Inputs:
# - resolved_accessions.tsv
# - REF.fasta
#
# Also consider these 4 accessions (duplicates removed):
# GCF_002291425.1, GCF_047901425.1, GCF_004342245.1, GCA_032062225.1
#
# Robustness:
# - Conda activation hook may reference JAVA_HOME under set -u (handled)
# - GFF validation ignores the ##FASTA FASTA block (valid GFF3)
# - FIXED: No more double Roary directories (script no longer pre-creates -f dir)
# Logs go to WORKDIR/logs and are also copied into the final Roary dir.
#
# Outputs:
# ${WORKDIR}/plot/core_tree.pdf
# ${WORKDIR}/plot/core_tree.png
###############################################################################
THREADS="${THREADS:-8}"
WORKDIR="${WORKDIR:-work_wgs_tree}"
RESOLVED_TSV="${RESOLVED_TSV:-resolved_accessions.tsv}"
REF_FASTA="${REF_FASTA:-AN6.fasta}"
ENV_NAME="${ENV_NAME:-/home/jhuang/miniconda3/envs/bengal3_ac3}"
EXTRA_ASSEMBLIES=(
#"GCF_002291425.1"
#"GCF_047901425.1"
#"GCF_004342245.1"
#"GCA_032062225.1"
)
CLUSTERS_K="${CLUSTERS_K:-6}"
MODE="${1:-all}"
log(){ echo "[$(date +'%F %T')] $*" >&2; }
need_cmd(){ command -v "$1" >/dev/null 2>&1; }
activate_existing_env(){
if [[ ! -d "${ENV_NAME}" ]]; then
log "ERROR: ENV_NAME path does not exist: ${ENV_NAME}"
exit 1
fi
conda_base="$(dirname "$(dirname "${ENV_NAME}")")"
if [[ -f "${conda_base}/etc/profile.d/conda.sh" ]]; then
# shellcheck disable=SC1091
source "${conda_base}/etc/profile.d/conda.sh"
else
if need_cmd conda; then
# shellcheck disable=SC1091
source "$(conda info --base)/etc/profile.d/conda.sh"
else
log "ERROR: cannot find conda.sh and conda is not on PATH."
exit 1
fi
fi
# Avoid "unbound variable" in activation hooks under set -u
export JAVA_HOME="${JAVA_HOME:-}"
log "Activating env: ${ENV_NAME}"
set +u
conda activate "${ENV_NAME}"
set -u
}
check_dependencies() {
# ---- plot-only mode: only need R (and optionally python) ----
if [[ "${MODE}" == "plot-only" ]]; then
local missing=()
command -v Rscript >/dev/null 2>&1 || missing+=("Rscript")
command -v python >/dev/null 2>&1 || missing+=("python")
if (( ${#missing[@]} )); then
log "ERROR: Missing required tools for plot-only in env: ${ENV_NAME}"
printf ' - %s\n' "${missing[@]}" >&2
exit 1
fi
# Check required R packages (fail early with clear message)
Rscript -e 'pkgs <- c("ggtree","ggplot2","aplot");
miss <- pkgs[!sapply(pkgs, requireNamespace, quietly=TRUE)];
if(length(miss)) stop("Missing R packages: ", paste(miss, collapse=", "))'
return 0
fi
# ------------------------------------------------------------
# existing full-pipeline checks continue below...
# (your current prokka/roary/raxml-ng checks stay as-is)
#...
}
prepare_accessions(){
[[ -s "${RESOLVED_TSV}" ]] || { log "ERROR: missing ${RESOLVED_TSV}"; exit 1; }
mkdir -p "${WORKDIR}/meta"
printf "%s\n" "${EXTRA_ASSEMBLIES[@]}" > "${WORKDIR}/meta/extras.txt"
WORKDIR="${WORKDIR}" RESOLVED_TSV="${RESOLVED_TSV}" python - << 'PY'
import os
import pandas as pd
import pathlib
workdir = pathlib.Path(os.environ.get("WORKDIR", "work_wgs_tree"))
resolved_tsv = os.environ.get("RESOLVED_TSV", "resolved_accessions.tsv")
df = pd.read_csv(resolved_tsv, sep="\t")
# Expect columns like: label, best_accession (but be tolerant)
if "best_accession" not in df.columns:
df = df.rename(columns={df.columns[1]:"best_accession"})
if "label" not in df.columns:
df = df.rename(columns={df.columns[0]:"label"})
df = df[["label","best_accession"]].dropna()
df = df[df["best_accession"]!="NA"].copy()
extras_path = workdir/"meta/extras.txt"
extras = [x.strip() for x in extras_path.read_text().splitlines() if x.strip()]
extra_df = pd.DataFrame({"label":[f"EXTRA_{a}" for a in extras], "best_accession": extras})
all_df = pd.concat([df, extra_df], ignore_index=True)
all_df = all_df.drop_duplicates(subset=["best_accession"], keep="first").reset_index(drop=True)
out = workdir/"meta/accessions.tsv"
out.parent.mkdir(parents=True, exist_ok=True)
all_df.to_csv(out, sep="\t", index=False)
print("Final unique genomes:", len(all_df))
print(all_df)
print("Wrote:", out)
PY
}
download_genomes(){
mkdir -p "${WORKDIR}/genomes_ncbi"
while IFS=$'\t' read -r label acc; do
[[ "$label" == "label" ]] && continue
[[ -z "${acc}" ]] && continue
outdir="${WORKDIR}/genomes_ncbi/${acc}"
if [[ -d "${outdir}" ]]; then
log "Found ${acc}, skipping download"
continue
fi
log "Downloading ${acc}..."
datasets download genome accession "${acc}" --include genome --filename "${WORKDIR}/genomes_ncbi/${acc}.zip"
unzip -q "${WORKDIR}/genomes_ncbi/${acc}.zip" -d "${outdir}"
rm -f "${WORKDIR}/genomes_ncbi/${acc}.zip"
done < "${WORKDIR}/meta/accessions.tsv"
}
collect_fastas(){
mkdir -p "${WORKDIR}/fastas"
while IFS=$'\t' read -r label acc; do
[[ "$label" == "label" ]] && continue
[[ -z "${acc}" ]] && continue
fna="$(find "${WORKDIR}/genomes_ncbi/${acc}" -type f -name "*.fna" | head -n 1 || true)"
[[ -n "${fna}" ]] || { log "ERROR: .fna not found for ${acc}"; exit 1; }
cp -f "${fna}" "${WORKDIR}/fastas/${acc}.fna"
done < "${WORKDIR}/meta/accessions.tsv"
[[ -s "${REF_FASTA}" ]] || { log "ERROR: missing ${REF_FASTA}"; exit 1; }
cp -f "${REF_FASTA}" "${WORKDIR}/fastas/REF.fna"
}
run_prokka(){
mkdir -p "${WORKDIR}/prokka" "${WORKDIR}/gffs"
for fna in "${WORKDIR}/fastas/"*.fna; do
base="$(basename "${fna}" .fna)"
outdir="${WORKDIR}/prokka/${base}"
gffout="${WORKDIR}/gffs/${base}.gff"
if [[ -s "${gffout}" ]]; then
log "GFF exists for ${base}, skipping Prokka"
continue
fi
log "Prokka annotating ${base}..."
prokka --outdir "${outdir}" --prefix "${base}" --cpus "${THREADS}" --force "${fna}"
cp -f "${outdir}/${base}.gff" "${gffout}"
done
}
sanitize_and_check_gffs(){
log "Sanity checking GFFs (ignoring ##FASTA section)..."
for gff in "${WORKDIR}/gffs/"*.gff; do
if file "$gff" | grep -qi "CRLF"; then
log "Fixing CRLF -> LF in $(basename "$gff")"
sed -i 's/\r$//' "$gff"
fi
bad=$(awk '
BEGIN{bad=0; in_fasta=0}
/^##FASTA/{in_fasta=1; next}
in_fasta==1{next}
/^#/{next}
NF==0{next}
{
if (split($0,a,"\t")!=9) {bad=1}
}
END{print bad}
' "$gff")
if [[ "$bad" == "1" ]]; then
log "ERROR: GFF feature section not 9-column tab-delimited: $gff"
log "First 5 problematic feature lines (before ##FASTA):"
awk '
BEGIN{in_fasta=0; c=0}
/^##FASTA/{in_fasta=1; next}
in_fasta==1{next}
/^#/{next}
NF==0{next}
{
if (split($0,a,"\t")!=9) {
print
c++
if (c==5) exit
}
}
' "$gff" || true
exit 1
fi
done
}
run_roary(){
mkdir -p "${WORKDIR}/meta" "${WORKDIR}/logs"
ts="$(date +%s)"
run_id="${ts}_$$"
ROARY_OUT="${WORKDIR}/roary_${run_id}"
ROARY_STDOUT="${WORKDIR}/logs/roary_${run_id}.stdout.txt"
ROARY_STDERR="${WORKDIR}/logs/roary_${run_id}.stderr.txt"
MARKER="${WORKDIR}/meta/roary_${run_id}.start"
: > "${MARKER}"
log "Running Roary (outdir: ${ROARY_OUT})"
log "Roary logs:"
log " STDOUT: ${ROARY_STDOUT}"
log " STDERR: ${ROARY_STDERR}"
set +e
roary -e --mafft -p "${THREADS}" -cd 95 -i 95 \
-f "${ROARY_OUT}" "${WORKDIR}/gffs/"*.gff \
> "${ROARY_STDOUT}" 2> "${ROARY_STDERR}"
rc=$?
set -e
if [[ "${rc}" -ne 0 ]]; then
log "WARNING: Roary exited non-zero (rc=${rc}). Will check if core alignment was produced anyway."
fi
CORE_ALN="$(find "${WORKDIR}" -maxdepth 2 -type f -name "core_gene_alignment.aln" -newer "${MARKER}" -printf '%T@ %p\n' 2>/dev/null \
| sort -nr | head -n 1 | cut -d' ' -f2- || true)"
if [[ -z "${CORE_ALN}" || ! -s "${CORE_ALN}" ]]; then
log "ERROR: Could not find core_gene_alignment.aln produced by this Roary run under ${WORKDIR}"
log "---- STDERR (head) ----"
head -n 120 "${ROARY_STDERR}" 2>/dev/null || true
log "---- STDERR (tail) ----"
tail -n 120 "${ROARY_STDERR}" 2>/dev/null || true
exit 1
fi
CORE_DIR="$(dirname "${CORE_ALN}")"
cp -f "${ROARY_STDOUT}" "${CORE_DIR}/roary.stdout.txt" || true
cp -f "${ROARY_STDERR}" "${CORE_DIR}/roary.stderr.txt" || true
# >>> IMPORTANT FIX: store ABSOLUTE path <<<
CORE_ALN_ABS="$(readlink -f "${CORE_ALN}")"
log "Using core alignment: ${CORE_ALN_ABS}"
echo "${CORE_ALN_ABS}" > "${WORKDIR}/meta/core_alignment_path.txt"
echo "$(readlink -f "${CORE_DIR}")" > "${WORKDIR}/meta/roary_output_dir.txt"
}
run_raxmlng(){
mkdir -p "${WORKDIR}/raxmlng"
CORE_ALN="$(cat "${WORKDIR}/meta/core_alignment_path.txt")"
[[ -s "${CORE_ALN}" ]] || { log "ERROR: core alignment not found or empty: ${CORE_ALN}"; exit 1; }
log "Running RAxML-NG..."
raxml-ng --all \
--msa "${CORE_ALN}" \
--model GTR+G \
--bs-trees 1000 \
--threads "${THREADS}" \
--prefix "${WORKDIR}/raxmlng/core"
}
ensure_r_pkgs(){
Rscript - <<'RS'
need <- c("ape","ggplot2","dplyr","readr","aplot","ggtree")
missing <- need[!vapply(need, requireNamespace, logical(1), quietly=TRUE)]
if (length(missing)) {
message("Missing R packages: ", paste(missing, collapse=", "))
message("Try:")
message(" conda install -c conda-forge -c bioconda r-aplot bioconductor-ggtree r-ape r-ggplot2 r-dplyr r-readr")
quit(status=1)
}
RS
}
plot_tree(){
mkdir -p "${WORKDIR}/plot"
WORKDIR="${WORKDIR}" python - << 'PY'
import os
import pandas as pd
import pathlib
workdir = pathlib.Path(os.environ.get("WORKDIR", "work_wgs_tree"))
acc = pd.read_csv(workdir/"meta/accessions.tsv", sep="\t")
g = (acc.groupby("best_accession")["label"]
.apply(lambda x: "; ".join(sorted(set(map(str, x)))))
.reset_index())
g["display"] = g.apply(lambda r: f'{r["label"]} ({r["best_accession"]})', axis=1)
labels = g.rename(columns={"best_accession":"sample"})[["sample","display"]]
# Add REF
labels = pd.concat([labels, pd.DataFrame([{"sample":"REF","display":"REF"}])], ignore_index=True)
out = workdir/"plot/labels.tsv"
out.parent.mkdir(parents=True, exist_ok=True)
labels.to_csv(out, sep="\t", index=False)
print("Wrote:", out)
PY
cat > "${WORKDIR}/plot/plot_tree.R" << 'RS'
suppressPackageStartupMessages({
library(ape); library(ggplot2); library(ggtree); library(dplyr); library(readr)
})
args <- commandArgs(trailingOnly=TRUE)
tree_in <- args[1]; labels_tsv <- args[2]; k <- as.integer(args[3])
out_pdf <- args[4]; out_png <- args[5]
tr <- read.tree(tree_in)
lab <- read_tsv(labels_tsv, show_col_types=FALSE)
tipmap <- setNames(lab$display, lab$sample)
tr$tip.label <- ifelse(tr$tip.label %in% names(tipmap), tipmap[tr$tip.label], tr$tip.label)
hc <- as.hclust.phylo(tr)
grp <- cutree(hc, k=k)
grp_df <- tibble(tip=names(grp), clade=paste0("Clade_", grp))
p <- ggtree(tr, layout="rectangular") %<+% grp_df +
aes(color=clade) +
geom_tree(linewidth=0.9) +
geom_tippoint(aes(color=clade), size=2.3) +
geom_tiplab(aes(color=clade), size=3.1, align=TRUE,
linetype="dotted", linesize=0.35, offset=0.02) +
theme_tree2() +
theme(legend.position="right", legend.title=element_blank(),
plot.margin=margin(8,18,8,8))
# + geom_treescale(x=0, y=0, width=0.01, fontsize=3)
# ---- Manual scale bar (fixed label "0.01") ----
scale_x <- 0
scale_y <- 0
scale_w <- 0.01
p <- p +
annotate("segment",
x = scale_x, xend = scale_x + scale_w,
y = scale_y, yend = scale_y,
linewidth = 0.6) +
annotate("text",
x = scale_x + scale_w/2,
y = scale_y - 0.6,
label = "0.01",
size = 3)
# ----------------------------------------------
ggsave(out_pdf, p, width=9, height=6.5, device="pdf")
ggsave(out_png, p, width=9, height=6.5, dpi=300)
RS
Rscript "${WORKDIR}/plot/plot_tree.R" \
"${WORKDIR}/raxmlng/core.raxml.support" \
"${WORKDIR}/plot/labels.tsv" \
"${CLUSTERS_K}" \
"${WORKDIR}/plot/core_tree.pdf" \
"${WORKDIR}/plot/core_tree.png"
log "Plot written:"
log " ${WORKDIR}/plot/core_tree.pdf"
log " ${WORKDIR}/plot/core_tree.png"
}
main(){
mkdir -p "${WORKDIR}"
activate_existing_env
check_dependencies
if [[ "${MODE}" == "plot-only" ]]; then
log "Running plot-only mode"
plot_tree
log "DONE."
exit 0
fi
log "1) Prepare unique accessions"
prepare_accessions
log "2) Download genomes"
download_genomes
log "3) Collect FASTAs (+ REF)"
collect_fastas
log "4) Prokka"
run_prokka
log "4b) GFF sanity check"
sanitize_and_check_gffs
log "5) Roary"
run_roary
log "6) RAxML-NG"
run_raxmlng
#log "6b) Check R packages"
#ensure_r_pkgs
log "7) Plot"
plot_tree
log "DONE."
}
main "$@"regenerate_labels.sh
python - <<'PY'
import json, re
from pathlib import Path
import pandas as pd
WORKDIR = Path("work_wgs_tree")
ACC_TSV = WORKDIR / "meta/accessions.tsv"
GENOMES_DIR = WORKDIR / "genomes_ncbi"
OUT = WORKDIR / "plot/labels.tsv"
def first_existing(paths):
for p in paths:
if p and Path(p).exists():
return Path(p)
return None
def find_metadata_files(acc_dir: Path):
# NCBI Datasets layouts vary by version; search broadly
candidates = []
for pat in [
"**/assembly_data_report.jsonl",
"**/data_report.jsonl",
"**/dataset_catalog.json",
"**/*assembly_report*.txt",
"**/*assembly_report*.tsv",
]:
candidates += list(acc_dir.glob(pat))
# de-dup, keep stable order
seen = set()
uniq = []
for p in candidates:
if p.as_posix() not in seen:
uniq.append(p)
seen.add(p.as_posix())
return uniq
def parse_jsonl_for_name_and_strain(p: Path):
# assembly_data_report.jsonl / data_report.jsonl: first JSON object usually has organism info
try:
with p.open() as f:
for line in f:
line = line.strip()
if not line:
continue
obj = json.loads(line)
# Try common fields
# organismName may appear as:
# obj["organism"]["organismName"] or obj["organismName"]
org = None
strain = None
if isinstance(obj, dict):
if "organism" in obj and isinstance(obj["organism"], dict):
org = obj["organism"].get("organismName") or obj["organism"].get("taxName")
# isolate/strain can hide in infraspecificNames or isolate/strain keys
infra = obj["organism"].get("infraspecificNames") or {}
if isinstance(infra, dict):
strain = infra.get("strain") or infra.get("isolate")
strain = strain or obj["organism"].get("strain") or obj["organism"].get("isolate")
org = org or obj.get("organismName") or obj.get("taxName")
# Sometimes isolate/strain is nested elsewhere
if not strain:
# assemblyInfo / assembly / sampleInfo patterns
for key in ["assemblyInfo", "assembly", "sampleInfo", "biosample"]:
if key in obj and isinstance(obj[key], dict):
d = obj[key]
strain = strain or d.get("strain") or d.get("isolate")
infra = d.get("infraspecificNames")
if isinstance(infra, dict):
strain = strain or infra.get("strain") or infra.get("isolate")
if org:
return org, strain
except Exception:
pass
return None, None
def parse_dataset_catalog(p: Path):
# dataset_catalog.json can include assembly/organism info, but structure varies.
try:
obj = json.loads(p.read_text())
except Exception:
return None, None
org = None
strain = None
# walk dict recursively looking for likely keys
def walk(x):
nonlocal org, strain
if isinstance(x, dict):
# organism keys
if not org:
if "organismName" in x and isinstance(x["organismName"], str):
org = x["organismName"]
elif "taxName" in x and isinstance(x["taxName"], str):
org = x["taxName"]
# strain/isolate keys
if not strain:
for k in ["strain", "isolate"]:
if k in x and isinstance(x[k], str) and x[k].strip():
strain = x[k].strip()
break
for v in x.values():
walk(v)
elif isinstance(x, list):
for v in x:
walk(v)
walk(obj)
return org, strain
def parse_assembly_report_txt(p: Path):
# NCBI assembly_report.txt often has lines like: "# Organism name:" and "# Infraspecific name:"
org = None
strain = None
try:
for line in p.read_text(errors="ignore").splitlines():
if line.startswith("# Organism name:"):
org = line.split(":", 1)[1].strip()
elif line.startswith("# Infraspecific name:"):
val = line.split(":", 1)[1].strip()
# e.g. "strain=XXXX" or "isolate=YYYY"
m = re.search(r"(strain|isolate)\s*=\s*(.+)", val)
if m:
strain = m.group(2).strip()
if org and strain:
break
except Exception:
pass
return org, strain
def best_name_for_accession(acc: str):
acc_dir = GENOMES_DIR / acc
if not acc_dir.exists():
return None
files = find_metadata_files(acc_dir)
org = None
strain = None
# Prefer JSONL reports first
for p in files:
if p.name.endswith(".jsonl"):
org, strain = parse_jsonl_for_name_and_strain(p)
if org:
break
# Next try dataset_catalog.json
if not org:
for p in files:
if p.name == "dataset_catalog.json":
org, strain = parse_dataset_catalog(p)
if org:
break
# Finally try assembly report text
if not org:
for p in files:
if "assembly_report" in p.name and p.suffix in [".txt", ".tsv"]:
org, strain = parse_assembly_report_txt(p)
if org:
break
if not org:
return None
# normalize whitespace
org = re.sub(r"\s+", " ", org).strip()
if strain:
strain = re.sub(r"\s+", " ", str(strain)).strip()
# avoid duplicating if strain already in organism string
if strain and strain.lower() not in org.lower():
return f"{org} {strain}"
return org
# --- build labels ---
acc = pd.read_csv(ACC_TSV, sep="\t")
if "label" not in acc.columns or "best_accession" not in acc.columns:
raise SystemExit("accessions.tsv must have columns: label, best_accession")
rows = []
for _, r in acc.iterrows():
label = str(r["label"])
accn = str(r["best_accession"])
if label.startswith("EXTRA_"):
nm = best_name_for_accession(accn)
if nm:
label = nm
else:
# fallback: keep previous behavior if metadata not found
label = label.replace("EXTRA_", "EXTRA ")
display = f"{label} ({accn})"
rows.append({"sample": accn, "display": display})
# Add GE11174 exactly as-is
rows.append({"sample": "GE11174", "display": "GE11174"})
out_df = pd.DataFrame(rows).drop_duplicates(subset=["sample"], keep="first")
OUT.parent.mkdir(parents=True, exist_ok=True)
out_df.to_csv(OUT, sep="\t", index=False)
print("Wrote:", OUT)
print(out_df)
PYplot_tree_v4.R
suppressPackageStartupMessages({
library(ape)
library(readr)
})
args <- commandArgs(trailingOnly = TRUE)
tree_in <- args[1]
labels_tsv <- args[2]
# args[3] is k (ignored here since all-black)
out_pdf <- args[4]
out_png <- args[5]
# --- Load tree ---
tr <- read.tree(tree_in)
# --- Root on outgroup (Brenneria nigrifluens) by accession ---
outgroup_id <- "GCF_005484965.1"
if (outgroup_id %in% tr$tip.label) {
tr <- root(tr, outgroup = outgroup_id, resolve.root = TRUE)
} else {
warning("Outgroup tip not found in tree: ", outgroup_id, " (tree will remain unrooted)")
}
# Make plotting order nicer
tr <- ladderize(tr, right = FALSE)
# --- Load labels (columns: sample, display) ---
lab <- read_tsv(labels_tsv, show_col_types = FALSE)
if (!all(c("sample","display") %in% colnames(lab))) {
stop("labels.tsv must contain columns: sample, display")
}
# Map tip labels AFTER rooting (rooting uses accession IDs)
tipmap <- setNames(lab$display, lab$sample)
tr$tip.label <- ifelse(tr$tip.label %in% names(tipmap),
unname(tipmap[tr$tip.label]),
tr$tip.label)
# --- Plot helper ---
plot_one <- function(device_fun) {
device_fun()
op <- par(no.readonly = TRUE)
on.exit(par(op), add = TRUE)
# Bigger right margin for long labels; tighter overall
par(mar = c(4, 2, 2, 18), xpd = NA)
# Compute xlim with padding so labels fit but whitespace is limited
xx <- node.depth.edgelength(tr)
xmax <- max(xx)
xpad <- 0.10 * xmax
plot(tr,
type = "phylogram",
use.edge.length = TRUE,
show.tip.label = TRUE,
edge.color = "black",
tip.color = "black",
cex = 0.9, # smaller text -> less overlap
label.offset = 0.003, # small gap after tip
no.margin = FALSE,
x.lim = c(0, xmax + xpad))
# Add a clear scale bar near bottom-left
# Use a fixed fraction of tree length for bar length
bar_len <- 0.05 * xmax
add.scale.bar(x = 0, y = 0, length = 0.01, lwd = 2, cex = 0.9)
}
# --- Write outputs (shorter height -> less vertical whitespace) ---
plot_one(function() pdf(out_pdf, width = 11, height = 6, useDingbats = FALSE))
dev.off()
plot_one(function() png(out_png, width = 3000, height = 1000, res = 300))
dev.off()
cat("Wrote:\n", out_pdf, "\n", out_png, "\n", sep = "")run_fastani_batch_verbose.sh
#!/usr/bin/env bash
set -euo pipefail
# ============ CONFIG ============
QUERY="bacass_out/Prokka/An6/An6.fna" # 你的 query fasta
ACC_LIST="accessions.txt" # 每行一个 GCF/GCA
OUTDIR="fastani_batch"
THREADS=8
SUFFIX=".genomic.fna"
# =================================
ts() { date +"%F %T"; }
log() { echo "[$(ts)] $*"; }
die() { echo "[$(ts)] ERROR: $*" >&2; exit 1; }
# --- checks ---
log "Checking required commands..."
for cmd in fastANI awk sort unzip find grep wc head readlink; do
command -v "$cmd" >/dev/null 2>&1 || die "Missing command: $cmd"
done
command -v datasets >/dev/null 2>&1 || die "Missing NCBI datasets CLI. Install from NCBI Datasets."
[[ -f "$QUERY" ]] || die "QUERY not found: $QUERY"
[[ -f "$ACC_LIST" ]] || die "Accession list not found: $ACC_LIST"
log "QUERY: $QUERY"
log "ACC_LIST: $ACC_LIST"
log "OUTDIR: $OUTDIR"
log "THREADS: $THREADS"
mkdir -p "$OUTDIR/ref_fasta" "$OUTDIR/zips" "$OUTDIR/tmp" "$OUTDIR/logs"
REF_LIST="$OUTDIR/ref_list.txt"
QUERY_LIST="$OUTDIR/query_list.txt"
RAW_OUT="$OUTDIR/fastani_raw.tsv"
FINAL_OUT="$OUTDIR/fastani_results.tsv"
DL_LOG="$OUTDIR/logs/download.log"
ANI_LOG="$OUTDIR/logs/fastani.log"
: > "$REF_LIST"
: > "$DL_LOG"
: > "$ANI_LOG"
# --- build query list ---
q_abs="$(readlink -f "$QUERY")"
echo "$q_abs" > "$QUERY_LIST"
log "Wrote query list: $QUERY_LIST"
log " -> $q_abs"
# --- download refs ---
log "Downloading reference genomes via NCBI datasets..."
n_ok=0
n_skip=0
while read -r acc; do
[[ -z "$acc" ]] && continue
[[ "$acc" =~ ^# ]] && continue
log "Ref: $acc"
zip="$OUTDIR/zips/${acc}.zip"
unpack="$OUTDIR/tmp/$acc"
out_fna="$OUTDIR/ref_fasta/${acc}${SUFFIX}"
# download zip
log " - datasets download -> $zip"
if datasets download genome accession "$acc" --include genome --filename "$zip" >>"$DL_LOG" 2>&1; then
log " - download OK"
else
log " - download FAILED (see $DL_LOG), skipping $acc"
n_skip=$((n_skip+1))
continue
fi
# unzip
rm -rf "$unpack"
mkdir -p "$unpack"
log " - unzip -> $unpack"
if unzip -q "$zip" -d "$unpack" >>"$DL_LOG" 2>&1; then
log " - unzip OK"
else
log " - unzip FAILED (see $DL_LOG), skipping $acc"
n_skip=$((n_skip+1))
continue
fi
# find genomic.fna (兼容不同包结构:优先找 genomic.fna,其次找任何 .fna)
fna="$(find "$unpack" -type f \( -name "*genomic.fna" -o -name "*genomic.fna.gz" \) | head -n 1 || true)"
if [[ -z "${fna:-}" ]]; then
log " - genomic.fna not found, try any *.fna"
fna="$(find "$unpack" -type f -name "*.fna" | head -n 1 || true)"
fi
if [[ -z "${fna:-}" ]]; then
log " - FAILED to find any .fna in package (see $DL_LOG). skipping $acc"
n_skip=$((n_skip+1))
continue
fi
# handle gz if needed
if [[ "$fna" == *.gz ]]; then
log " - found gzipped fasta: $(basename "$fna"), gunzip -> $out_fna"
gunzip -c "$fna" > "$out_fna"
else
log " - found fasta: $(basename "$fna"), copy -> $out_fna"
cp -f "$fna" "$out_fna"
fi
# sanity check fasta looks non-empty
if [[ ! -s "$out_fna" ]]; then
log " - output fasta is empty, skipping $acc"
n_skip=$((n_skip+1))
continue
fi
echo "$(readlink -f "$out_fna")" >> "$REF_LIST"
n_ok=$((n_ok+1))
log " - saved ref fasta OK"
done < "$ACC_LIST"
log "Download summary: OK=$n_ok, skipped=$n_skip"
log "Ref list written: $REF_LIST ($(wc -l < "$REF_LIST") refs)"
if [[ "$(wc -l < "$REF_LIST")" -eq 0 ]]; then
die "No references available. Check $DL_LOG"
fi
# --- run fastANI ---
log "Running fastANI..."
log "Command:"
log " fastANI -ql $QUERY_LIST -rl $REF_LIST -t $THREADS -o $RAW_OUT"
# 重要:不要吞掉错误信息,把 stdout/stderr 进日志
if fastANI -ql "$QUERY_LIST" -rl "$REF_LIST" -t "$THREADS" -o "$RAW_OUT" >>"$ANI_LOG" 2>&1; then
log "fastANI finished (see $ANI_LOG)"
else
log "fastANI FAILED (see $ANI_LOG)"
die "fastANI failed. Inspect $ANI_LOG"
fi
# --- verify raw output ---
if [[ ! -f "$RAW_OUT" ]]; then
die "fastANI did not create $RAW_OUT. Check $ANI_LOG"
fi
if [[ ! -s "$RAW_OUT" ]]; then
die "fastANI output is empty ($RAW_OUT). Check $ANI_LOG; also verify fasta validity."
fi
log "fastANI raw output: $RAW_OUT ($(wc -l < "$RAW_OUT") lines)"
log "Sample lines:"
head -n 5 "$RAW_OUT" || true
# --- create final table ---
log "Creating final TSV with header..."
echo -e "Query\tReference\tANI\tMatchedFrag\tTotalFrag" > "$FINAL_OUT"
awk 'BEGIN{OFS="\t"} {print $1,$2,$3,$4,$5}' "$RAW_OUT" >> "$FINAL_OUT"
log "Final results: $FINAL_OUT ($(wc -l < "$FINAL_OUT") lines incl. header)"
log "Top hits (ANI desc):"
tail -n +2 "$FINAL_OUT" | sort -k3,3nr | head -n 10 || true
log "DONE."
log "Logs:"
log " download log: $DL_LOG"
log " fastANI log: $ANI_LOG"