New Linkedin Link
Leave a reply
Environment Setup: It sets up a Conda environment named picrust2, using the conda create command and then activates this environment using conda activate picrust2.
#https://github.com/picrust/picrust2/wiki/PICRUSt2-Tutorial-(v2.2.0-beta)#minimum-requirements-to-run-full-tutorial
mamba create -n picrust2 -c bioconda -c conda-forge picrust2 #2.5.3 #=2.2.0_b
mamba activate /home/jhuang/miniconda3/envs/picrust2Under docker-env (qiime2-amplicon-2023.9)
Export QIIME2 feature table and representative sequences
#docker pull quay.io/qiime2/core:2023.9
#docker run -it --rm \
#-v /mnt/md1/DATA/Data_Karoline_16S_2025:/data \
#-v /home/jhuang/REFs:/home/jhuang/REFs \
#quay.io/qiime2/core:2023.9 bash
#cd /data
# === SETTINGS ===
FEATURE_TABLE_QZA="dada2_tests2/test_7_f240_r240/table.qza"
REP_SEQS_QZA="dada2_tests2/test_7_f240_r240/rep-seqs.qza"
# === STEP 1: EXPORT QIIME2 ARTIFACTS ===
mkdir -p qiime2_export
qiime tools export --input-path $FEATURE_TABLE_QZA --output-path qiime2_export
qiime tools export --input-path $REP_SEQS_QZA --output-path qiime2_exportConvert BIOM to TSV for Picrust2 input
biom convert \
-i qiime2_export/feature-table.biom \
-o qiime2_export/feature-table.tsv \
--to-tsvUnder env (picrust2): mamba activate /home/jhuang/miniconda3/envs/picrust2
Run PICRUSt2 pipeline
tail -n +2 qiime2_export/feature-table.tsv > qiime2_export/feature-table-fixed.tsv
picrust2_pipeline.py \
-s qiime2_export/dna-sequences.fasta \
-i qiime2_export/feature-table-fixed.tsv \
-o picrust2_out \
-p 100
#This will:
#* Place sequences in the reference tree (using EPA-NG),
#* Predict gene family abundances (e.g., EC, KO, PFAM, TIGRFAM),
#* Predict pathway abundances.
#In current PICRUSt2 (with picrust2_pipeline.py), you do not run hsp.py separately.
#Instead, picrust2_pipeline.py internally runs the HSP step for all functional categories automatically. It outputs all the prediction files (16S_predicted_and_nsti.tsv.gz, COG_predicted.tsv.gz, PFAM_predicted.tsv.gz, KO_predicted.tsv.gz, EC_predicted.tsv.gz, TIGRFAM_predicted.tsv.gz, PHENO_predicted.tsv.gz) in the output directory.
mkdir picrust2_out_advanced; cd picrust2_out_advanced
#If you still want to run hsp.py manually (advanced use / debugging), the commands correspond directly:
hsp.py -i 16S -t ../picrust2_out/out.tre -o 16S_predicted_and_nsti.tsv.gz -p 100 -n
hsp.py -i COG -t ../picrust2_out/out.tre -o COG_predicted.tsv.gz -p 100
hsp.py -i PFAM -t ../picrust2_out/out.tre -o PFAM_predicted.tsv.gz -p 100
hsp.py -i KO -t ../picrust2_out/out.tre -o KO_predicted.tsv.gz -p 100
hsp.py -i EC -t ../picrust2_out/out.tre -o EC_predicted.tsv.gz -p 100
hsp.py -i TIGRFAM -t ../picrust2_out/out.tre -o TIGRFAM_predicted.tsv.gz -p 100
hsp.py -i PHENO -t ../picrust2_out/out.tre -o PHENO_predicted.tsv.gz -p 100Metagenome prediction per functional category (if needed separately)
#cd picrust2_out_advanced
metagenome_pipeline.py -i ../qiime2_export/feature-table.biom -m 16S_predicted_and_nsti.tsv.gz -f COG_predicted.tsv.gz -o COG_metagenome_out --strat_out
metagenome_pipeline.py -i ../qiime2_export/feature-table.biom -m 16S_predicted_and_nsti.tsv.gz -f EC_predicted.tsv.gz -o EC_metagenome_out --strat_out
metagenome_pipeline.py -i ../qiime2_export/feature-table.biom -m 16S_predicted_and_nsti.tsv.gz -f KO_predicted.tsv.gz -o KO_metagenome_out --strat_out
metagenome_pipeline.py -i ../qiime2_export/feature-table.biom -m 16S_predicted_and_nsti.tsv.gz -f PFAM_predicted.tsv.gz -o PFAM_metagenome_out --strat_out
metagenome_pipeline.py -i ../qiime2_export/feature-table.biom -m 16S_predicted_and_nsti.tsv.gz -f TIGRFAM_predicted.tsv.gz -o TIGRFAM_metagenome_out --strat_out
# Add descriptions in gene family tables
add_descriptions.py -i COG_metagenome_out/pred_metagenome_unstrat.tsv.gz -m COG -o COG_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gz
add_descriptions.py -i EC_metagenome_out/pred_metagenome_unstrat.tsv.gz -m EC -o EC_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gz
add_descriptions.py -i KO_metagenome_out/pred_metagenome_unstrat.tsv.gz -m KO -o KO_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gz # EC and METACYC is a pair, EC for gene_annotation and METACYC for pathway_annotation
add_descriptions.py -i PFAM_metagenome_out/pred_metagenome_unstrat.tsv.gz -m PFAM -o PFAM_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gz
add_descriptions.py -i TIGRFAM_metagenome_out/pred_metagenome_unstrat.tsv.gz -m TIGRFAM -o TIGRFAM_metagenome_out/pred_metagenome_unstrat_descrip.tsv.gzPathway inference (MetaCyc pathways from EC numbers)
#cd picrust2_out_advanced
pathway_pipeline.py -i EC_metagenome_out/pred_metagenome_contrib.tsv.gz -o EC_pathways_out -p 100
pathway_pipeline.py -i EC_metagenome_out/pred_metagenome_unstrat.tsv.gz -o EC_pathways_out_per_seq -p 100 --per_sequence_contrib --per_sequence_abun EC_metagenome_out/seqtab_norm.tsv.gz --per_sequence_function EC_predicted.tsv.gz
#ERROR due to missing .../pathway_mapfiles/KEGG_pathways_to_KO.tsv
pathway_pipeline.py -i COG_metagenome_out/pred_metagenome_contrib.tsv.gz -o KEGG_pathways_out -p 100 --no_regroup --map /home/jhuang/anaconda3/envs/picrust2/lib/python3.6/site-packages/picrust2/default_files/pathway_mapfiles/KEGG_pathways_to_KO.tsv
pathway_pipeline.py -i KO_metagenome_out/pred_metagenome_strat.tsv.gz -o KEGG_pathways_out -p 100 --no_regroup --map /home/jhuang/anaconda3/envs/picrust2/lib/python3.6/site-packages/picrust2/default_files/pathway_mapfiles/KEGG_pathways_to_KO.tsv
add_descriptions.py -i EC_pathways_out/path_abun_unstrat.tsv.gz -m METACYC -o EC_pathways_out/path_abun_unstrat_descrip.tsv.gz
gunzip EC_pathways_out/path_abun_unstrat_descrip.tsv.gz
#Error - no rows remain after regrouping input table. The default pathway and regroup mapfiles are meant for EC numbers. Note that KEGG pathways are not supported since KEGG is a closed-source database, but you can input custom pathway mapfiles if you have access. If you are using a custom function database did you mean to set the --no-regroup flag and/or change the default pathways mapfile used?
#If ERROR --> USE the METACYC for downstream analyses!!!
#ERROR due to missing .../description_mapfiles/KEGG_pathways_info.tsv.gz
#add_descriptions.py -i KO_pathways_out/path_abun_unstrat.tsv.gz -o KEGG_pathways_out/path_abun_unstrat_descrip.tsv.gz --custom_map_table /home/jhuang/anaconda3/envs/picrust2/lib/python3.6/site-packages/picrust2/default_files/description_mapfiles/KEGG_pathways_info.tsv.gz
#NOTE: Target-analysis for the pathway "mixed acid fermentation"Visualization
#7.1 STAMP
#https://github.com/picrust/picrust2/wiki/STAMP-example
#Note that STAMP can only be opened under Windows
# It needs two files: path_abun_unstrat_descrip.tsv.gz as "Profile file" and metadata.tsv as "Group metadata file".
cp ~/DATA/Data_Karoline_16S_2025/picrust2_out_advanced/EC_pathways_out/path_abun_unstrat_descrip.tsv ~/DATA/Access_to_Win10/
cut -d$'\t' -f1 qiime2_metadata.tsv > 1
cut -d$'\t' -f3 qiime2_metadata.tsv > 3
cut -d$'\t' -f5-6 qiime2_metadata.tsv > 5_6
paste -d$'\t' 1 3 > 1_3
paste -d$'\t' 1_3 5_6 > metadata.tsv
#SampleID --> SampleID
SampleID Group pre_post Sex_age
sample-A1 Group1 3d.post.stroke male.aged
sample-A2 Group1 3d.post.stroke male.aged
sample-A3 Group1 3d.post.stroke male.aged
cp ~/DATA/Data_Karoline_16S_2025/metadata.tsv ~/DATA/Access_to_Win10/
# MANULLY_EDITING: keeping the only needed records in metadata.tsv: Group 9 (J1–J4, J10, J11) and Group 10 (K1–K6).
#7.2. ALDEx2
https://bioconductor.org/packages/release/bioc/html/ALDEx2.htmlUnder docker-env (qiime2-amplicon-2023.9)
(NOT_NEEDED) Convert pathway output to BIOM and re-import to QIIME2 gunzip picrust2_out/pathways_out/path_abun_unstrat.tsv.gz biom convert \ -i picrust2_out/pathways_out/path_abun_unstrat.tsv \ -o picrust2_out/path_abun_unstrat.biom \ –table-type=”Pathway table” \ –to-hdf5
qiime tools import \
--input-path picrust2_out/path_abun_unstrat.biom \
--type 'FeatureTable[Frequency]' \
--input-format BIOMV210Format \
--output-path path_abun.qza
#qiime tools export --input-path path_abun.qza --output-path exported_path_abun
#qiime tools peek path_abun.qza
echo "✅ PICRUSt2 pipeline complete. Output in: picrust2_out"Short answer: unless you had a very clear, pre-specified directional hypothesis, you should use a two-sided test.
A bit more detail:
* Two-sided t-test
* Tests: “Are the means different?” (could be higher or lower).
* Standard default in most biological and clinical studies and usually what reviewers expect.
* More conservative than a one-sided test.
* One-sided t-test
* Tests: “Is Group A greater than Group B?” (or strictly less than).
* You should only use it if before looking at the data you had a strong reason to expect a specific direction and you would ignore/consider uninterpretable a difference in the opposite direction.
* Using one-sided just to gain significance is considered bad practice.
For your pathway analysis (exploratory, many pathways, q-value correction), the safest and most defensible choice is to:
* Use a two-sided t-test (equal variance or Welch’s, depending on variance assumptions).
So I’d recommend rerunning STAMP with Type: Two-sided and reporting those results.
#--> Using a two-sided Welch's t-test in STAMP, that is the unequal-variance version (does not assume equal variances and is more conservative than “t-test (equal variance)” referring to the classical unpaired Student’s t-test.Statistics in STAMP
* For multiple groups:
* Statistical test: ANOVA, Kruskal-Wallis H-test
* Post-hoc test: Games-Howell, Scheffe, Tukey-Kramer, Welch's (uncorrected) (by default 0.95)
* Effect size: Eta-squared
* Multiple test correction: Benjamini-Hochberg FDR, Bonferroni, No correction
* For two groups
* Statistical test: t-test (equal variance), Welch's t-test, White's non-parametric t-test
* Type: One-sided, Two-sided
* CI method: "DP: Welch's inverted" (by default 0.95)
* Multiple test correction: Benjamini-Hochberg FDR, Bonferroni, No correction, Sidak, Storey FDR
* For two samples
* Statistical test: Bootstrap, Chi-square test, Chi-square test (w/Yates'), Difference between proportions, Fisher's exact test, G-test, G-test (w/Yates'), G-test (w/Yates') + Fisher's, Hypergeometric, Permutation
* Type: One-sided, Two-sided
* CI method: "DP: Asymptotic", "DP: Asymptotic-CC", "DP: Newcomber-Wilson", "DR: Haldane adjustment", "RP: Asymptotic" (by default 0.95)
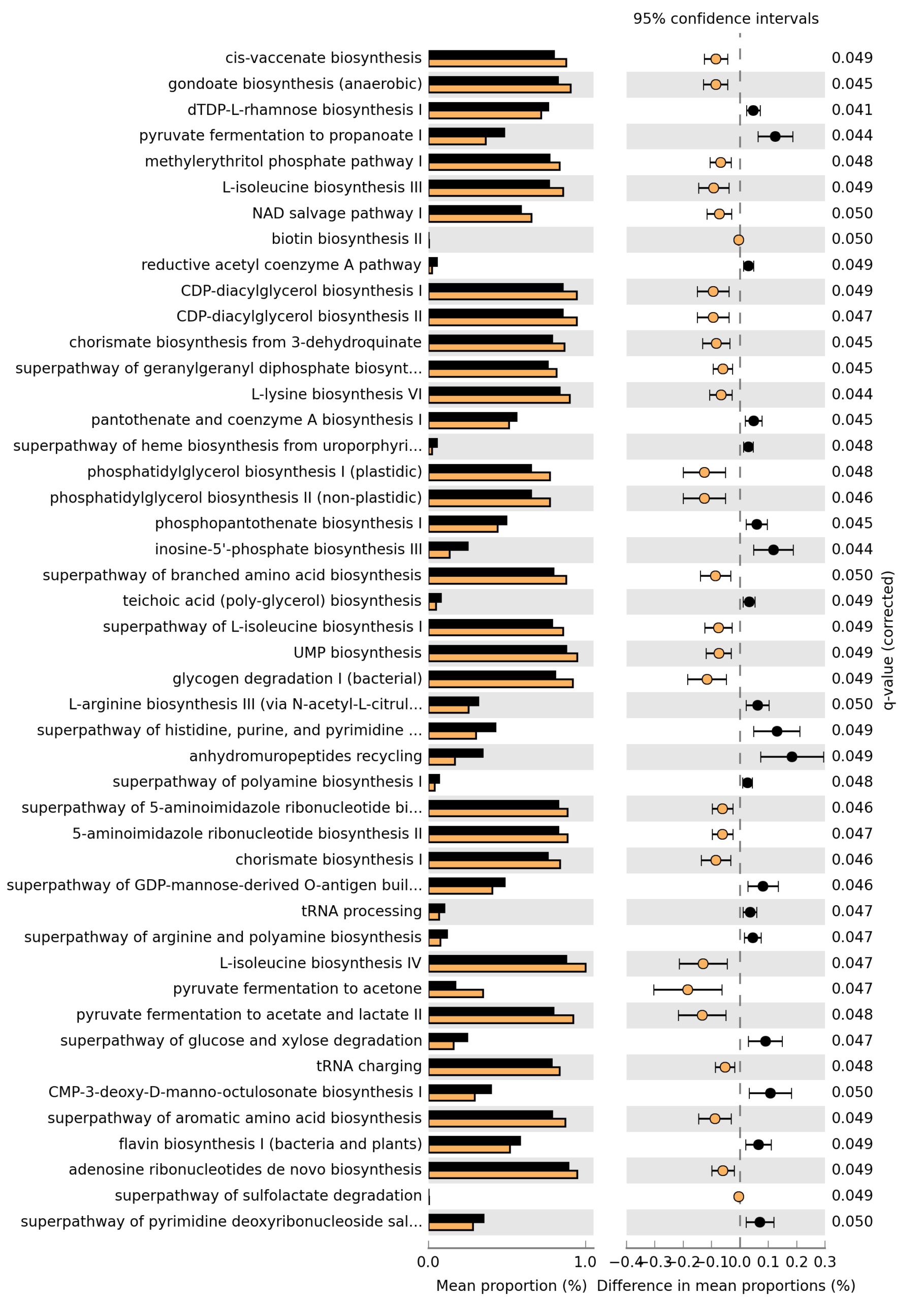
* Multiple test correction: Benjamini-Hochberg FDR, Bonferroni, No correction, Sidak, Storey FDRSince MetaCyc does not have a single pathway explicitly named “short-chain fatty acid biosynthesis”, I defined a small SCFA-related set (acetate-, propionate- and butyrate-producing pathways) and tested these between Group 9 and Group 10 (Welch’s t-test, with BH correction within this subset). These pathways can also be found in the file Welchs_t-test.xlsx attached to my email from 26.11.2025 (for Group9 (J1-4, J6-7, J10-11) vs Group10 (K1-6)).
Pathway ID Description Group 9 mean (%) Group 10 mean (%) p-value p-adj (BH, SCFA set)
P108-PWY pyruvate fermentation to propanoate I 0.5070 0.3817 0.001178 0.0071
PWY-5100 pyruvate fermentation to acetate and lactate II 0.8354 0.9687 0.007596 0.0228
CENTFERM-PWY pyruvate fermentation to butanoate 0.0766 0.0410 0.026608 0.0532
PWY-5677 succinate fermentation to butanoate 0.0065 0.0088 0.365051 0.5476
P163-PWY L-lysine fermentation to acetate and butanoate 0.0324 0.0271 0.484704 0.5816
PWY-5676 acetyl-CoA fermentation to butanoate II 0.1397 0.1441 0.927588 0.9276In this SCFA-focused set, the propionate (P108-PWY) and acetate (PWY-5100) pathways remain significantly different between Group 9 and Group 10 after adjustment, whereas the butyrate-related pathways do not show clear significant differences (CENTFERM-PWY is borderline).
from 14.01.2026 (for Group9 (J1-4, J10-11) vs Group10 (K1-6)), marked green in the Excel-files.
Pathway ID Description Group 9 mean (%) Group 10 mean (%) p-value p-adj (BH, 6-pathway set)
P108-PWY pyruvate fermentation to propanoate I 0.5142 0.3817 0.001354 0.008127
PWY-5100 pyruvate fermentation to acetate and lactate II 0.8401 0.9687 0.008763 0.026290
CENTFERM-PWY pyruvate fermentation to butanoate 0.0729 0.0410 0.069958 0.139916
PWY-5677 succinate fermentation to butanoate 0.0063 0.0088 0.367586 0.551379
P163-PWY L-lysine fermentation to acetate and butanoate 0.0308 0.0271 0.693841 0.832609
PWY-5676 acetyl-CoA fermentation to butanoate II 0.1421 0.1441 0.971290 0.971290Reporting
Please find attached the results of the pathway analysis. The Excel file contains the full statistics for all pathways; those with adjusted p-values (Benjamini–Hochberg) ≤ 0.05 are highlighted in yellow and are the ones illustrated in the figure.
The analysis was performed using Welch’s t-test (two-sided) with Benjamini–Hochberg correction for multiple testing.alleleexpression, cageseq, circrna, denovotranscript, differentialabundance, drop, dualrnaseq, evexplorer, isoseq, lncpipe, nanostring, nascent, rnafusion, rnaseq, rnasplice, rnavar, riboseq, slamseq, stableexpression
smrnaseq
marsseq, scdownstream, scflow, scnanoseq, scrnaseq, smartseq2
molkart, panoramaseq, pixelator, sopa, spatialvi, spatialxe
atacseq, callingcards, chipseq, clipseq, cutandrun, hic, hicar, mnaseseq, sammyseq, tfactivity
methylarray, methylong, methylseq
abotyper, circdna, deepvariant, eager, exoseq, gwas, longraredisease, mitodetect, oncoanalyser, pacvar, phaseimpute, radseq, raredisease, rarevariantburden, rnadnavar, sarek, ssds, tumourevo, variantbenchmarking, variantcatalogue, variantprioritization, createpanelrefs
pathogensurveillance, phageannotator, tbanalyzer, viralmetagenome, viralintegration, viralrecon, vipr
ampliseq, coproid, createtaxdb, detaxizer, funcscan, mag, magmap, metapep, metatdenovo, taxprofiler
bacass, bactmap, denovohybrid, genomeannotator, genomeassembler, genomeqc, genomeskim, hgtseq, multiplesequencealign, neutronstar, pangenome, pairgenomealign, phyloplace, reportho
airrflow, epitopeprediction, hlatyping, mhcquant
ddamsproteomics, diaproteomics, kmermaid, metaboigniter, proteinannotator, proteinfamilies, proteinfold, proteogenomicsdb, proteomicslfq, quantms, ribomsqc
cellpainting, imcyto, liverctanalysis, lsmquant, mcmicro, rangeland, troughgraph
bamtofastq, datasync, demo, demultiplex, fastqrepair, fastquorum, fetchngs, nanoseq, readsimulator, references, seqinspector, seqsubmit
crisprseq, crisprvar
deepmodeloptim, deepmutscan, diseasemodulediscovery, drugresponseeval, meerpipe, omicsgenetraitassociation, spinningjenny
| Category | Name | Short description | 中文描述 | Released | Stars | Last release |
|---|---|---|---|---|---|---|
| Bulk RNA-seq & transcriptomics | alleleexpression | Allele-specific expression (ASE) analysis using STAR-WASP, UMI-tools, phaser | 等位基因特异性表达(ASE)分析:STAR-WASP 比对,UMI-tools 去重,phaser 单倍型分相与 ASE 检测 | 2 | – | |
| Bulk RNA-seq & transcriptomics | cageseq | CAGE-sequencing analysis pipeline with trimming, alignment and counting of CAGE tags. | CAGE-seq 分析:剪切、比对并统计 CAGE 标签(转录起始相关)。 | 11 | 1.0.2 | |
| Bulk RNA-seq & transcriptomics | circrna | circRNA quantification, differential expression analysis and miRNA target prediction of RNA-Seq data | 环状 RNA(circRNA)定量、差异表达分析及 miRNA 靶标预测。 | 59 | – | |
| Bulk RNA-seq & transcriptomics | denovotranscript | de novo transcriptome assembly of paired-end short reads from bulk RNA-seq | 基于 bulk RNA-seq 双端短读长的从头转录组组装。 | 19 | 1.2.1 | |
| Bulk RNA-seq & transcriptomics | differentialabundance | Differential abundance analysis for feature/observation matrices (e.g., RNA-seq) | 对特征/观测矩阵做差异丰度分析(可用于表达矩阵等)。 | 87 | 1.5.0 | |
| Bulk RNA-seq & transcriptomics | drop | Pipeline to find aberrant events in RNA-Seq data, useful for diagnosis of rare disorders | RNA-seq 异常事件检测流程(用于罕见病诊断等)。 | 7 | – | |
| Bulk RNA-seq & transcriptomics | dualrnaseq | Analysis of Dual RNA-seq data (host-pathogen interactions) | 宿主-病原双 RNA-seq 分析流程,用于研究宿主-病原相互作用。 | 25 | 1.0.0 | |
| Bulk RNA-seq & transcriptomics | evexplorer | Analyze RNA data from extracellular vesicles; QC, region detection, normalization, DRE | 胞外囊泡(EV)RNA 数据分析:质控、表达区域检测、归一化与差异 RNA 表达(DRE)。 | 1 | – | |
| Bulk RNA-seq & transcriptomics | isoseq | Genome annotation with PacBio Iso-Seq from raw subreads to FLNC and bed annotation | PacBio Iso-Seq 基因组注释:从 subreads 生成 FLNC 并产出 bed 注释。 | 50 | 2.0.0 | |
| Bulk RNA-seq & transcriptomics | lncpipe | Analysis of long non-coding RNAs from RNA-seq datasets (under development) | lncRNA(长链非编码 RNA)分析流程(开发中)。 | 34 | – | |
| Bulk RNA-seq & transcriptomics | nanostring | Analysis pipeline for Nanostring nCounter expression data. | Nanostring nCounter 表达数据分析流程。 | 16 | 1.3.1 | |
| Bulk RNA-seq & transcriptomics | nascent | Nascent Transcription Processing Pipeline | 新生转录(nascent RNA)处理与分析流程。 | 22 | 2.3.0 | |
| Bulk RNA-seq & transcriptomics | rnafusion | RNA-seq analysis pipeline for detection of gene-fusions | RNA-seq 融合基因检测流程。 | 170 | 4.0.0 | |
| Bulk RNA-seq & transcriptomics | rnaseq | RNA sequencing pipeline (STAR/RSEM/HISAT2/Salmon) with QC and counts | 常规 bulk RNA-seq 分析:比对/定量/计数与全面质控(多比对/定量器可选)。 | 1179 | 3.22.2 | |
| Bulk RNA-seq & transcriptomics | rnasplice | RNA-seq alternative splicing analysis | RNA-seq 可变剪接分析流程。 | 63 | 1.0.4 | |
| Bulk RNA-seq & transcriptomics | rnavar | gatk4 RNA variant calling pipeline | 基于 GATK4 的 RNA 变异检测(RNA variant calling)。 | 58 | 1.2.2 | |
| Bulk RNA-seq & transcriptomics | riboseq | Analysis of ribosome profiling (Ribo-seq) data | Ribo-seq(核糖体测序/核糖体 footprinting)分析流程。 | 21 | 1.2.0 | |
| Bulk RNA-seq & transcriptomics | slamseq | SLAMSeq processing and analysis pipeline | SLAM-seq(新生 RNA 标记)处理与分析流程。 | 10 | 1.0.0 | |
| Bulk RNA-seq & transcriptomics | stableexpression | Identify stable genes across datasets; useful for RT-qPCR reference genes | 寻找最稳定基因(适合作为 RT-qPCR 参考内参基因)。 | 5 | – | |
| Small RNA-seq | smrnaseq | A small-RNA sequencing analysis pipeline | 小 RNA 测序(如 miRNA 等)分析流程。 | 98 | 2.4.1 | |
| Single-cell transcriptomics | marsseq | MARS-seq v2 pre-processing pipeline with velocity | MARS-seq v2 预处理流程,支持 RNA velocity。 | 8 | 1.0.3 | |
| Single-cell transcriptomics | scdownstream | Single cell transcriptomics pipeline for QC, integration, presentation | 单细胞转录组下游:质控、整合与结果展示。 | 81 | – | |
| Single-cell transcriptomics | scflow | Please consider using/contributing to nf-core/scdownstream | 单细胞流程(建议转向/贡献 scdownstream)。 | 25 | – | |
| Single-cell transcriptomics | scnanoseq | Single-cell/nuclei pipeline for Oxford Nanopore + 10x Genomics | 单细胞/细胞核测序流程:结合 ONT 与 10x 数据。 | 52 | 1.2.1 | |
| Single-cell transcriptomics | scrnaseq | Single-cell RNA-Seq pipeline (10x/DropSeq/SmartSeq etc.) | 单细胞 RNA-seq 主流程:支持 10x、DropSeq、SmartSeq 等。 | 310 | 4.1.0 | |
| Single-cell transcriptomics | smartseq2 | Process single cell RNA-seq generated with SmartSeq2 | SmartSeq2 单细胞 RNA-seq 处理流程。 | 15 | – | |
| Spatial omics | molkart | Processing Molecular Cartography data (Resolve Bioscience combinatorial FISH) | Resolve Molecular Cartography(组合 FISH)数据处理流程。 | 14 | 1.2.0 | |
| Spatial omics | panoramaseq | Pipeline to process sequencing-based spatial transcriptomics data (in-situ arrays) | 测序型空间转录组(in-situ arrays)数据处理流程。 | 0 | – | |
| Spatial omics | pixelator | Pipeline to generate Molecular Pixelation data (Pixelgen) | Pixelgen 分子像素化(Molecular Pixelation)数据处理流程。 | 13 | 2.3.0 | |
| Spatial omics | sopa | Nextflow version of Sopa – spatial omics pipeline and analysis | Sopa 的 Nextflow 实现:空间组学流程与分析。 | 11 | – | |
| Spatial omics | spatialvi | Process spatial gene counts + spatial coordinates + image data (10x Visium) | 10x Visium 空间转录组处理:基因计数+空间坐标+图像数据。 | 70 | – | |
| Spatial omics | spatialxe | (no description shown) | 空间组学相关流程(原表未给出描述)。 | 24 | – | |
| Chromatin & regulation | atacseq | ATAC-seq peak-calling and QC analysis pipeline | ATAC-seq 峰识别与质控分析流程。 | 221 | 2.1.2 | |
| Chromatin & regulation | callingcards | A pipeline for processing calling cards data | Calling cards 实验数据处理流程。 | 6 | 1.0.0 | |
| Chromatin & regulation | chipseq | ChIP-seq peak-calling, QC and differential analysis | ChIP-seq 峰识别、质控与差异分析流程。 | 229 | 2.1.0 | |
| Chromatin & regulation | clipseq | CLIP-seq QC, mapping, UMI deduplication, peak-calling options | CLIP-seq 分析:质控、比对、UMI 去重与多种 peak calling。 | 24 | 1.0.0 | |
| Chromatin & regulation | cutandrun | CUT&RUN / CUT&TAG pipeline with QC, spike-ins, IgG controls, peak calling | CUT&RUN/CUT&TAG 分析:质控、spike-in、IgG 对照、峰识别与下游。 | 106 | 3.2.2 | |
| Chromatin & regulation | hic | Analysis of Chromosome Conformation Capture (Hi-C) data | Hi-C 染色体构象捕获数据分析流程。 | 105 | 2.1.0 | |
| Chromatin & regulation | hicar | HiCAR multi-omic co-assay pipeline | HiCAR 多组学共测(转录+染色质可及性+接触)分析流程。 | 12 | 1.0.0 | |
| Chromatin & regulation | mnaseseq | MNase-seq analysis pipeline using BWA and DANPOS2 | MNase-seq 分析流程(BWA + DANPOS2)。 | 12 | 1.0.0 | |
| Chromatin & regulation | sammyseq | SAMMY-seq pipeline to analyze chromatin state | SAMMY-seq 染色质状态分析流程。 | 5 | – | |
| Chromatin & regulation | tfactivity | Identify differentially active TFs using expression + open chromatin | 整合表达与开放染色质数据,识别差异活跃转录因子(TF)。 | 12 | – | |
| DNA methylation | methylarray | Illumina methylation array processing; QC, confounders, DMP/DMR, cell comp optional | Illumina 甲基化芯片分析:预处理、质控、混杂因素检查、DMP/DMR;可选细胞组成估计与校正。 | 6 | – | |
| DNA methylation | methylong | Extract methylation calls from long reads (ONT/PacBio) | 从长读长(ONT/PacBio)提取甲基化识别结果。 | 19 | 2.0.0 | |
| DNA methylation | methylseq | Bisulfite-seq methylation pipeline (Bismark/bwa-meth + MethylDackel/rastair) | 亚硫酸氢盐测序甲基化分析流程(Bismark/bwa-meth 等)。 | 185 | 4.2.0 | |
| Human genomics, variants & disease | abotyper | Characterise human blood group and red cell antigens using ONT | 基于 ONT 的人类血型与红细胞抗原分型/鉴定流程。 | 1 | – | |
| Human genomics, variants & disease | circdna | Identify extrachromosomal circular DNA (ecDNA) from Circle-seq/WGS/ATAC-seq | 从 Circle-seq/WGS/ATAC-seq 识别染色体外环状 DNA(ecDNA)。 | 31 | 1.1.0 | |
| Human genomics, variants & disease | createpanelrefs | Generate Panel of Normals / models / references from many samples | 从大量样本生成 PoN(Panel of Normals)/模型/参考资源。 | 11 | – | |
| Human genomics, variants & disease | deepvariant | Consider using/contributing to nf-core/sarek | DeepVariant 相关(建议使用/贡献至 sarek)。 | 40 | 1.0 | |
| Human genomics, variants & disease | eager | Ancient DNA analysis pipeline | 古 DNA(aDNA)分析流程(可重复、标准化)。 | 195 | 2.5.3 | |
| Human genomics, variants & disease | exoseq | Please consider using/contributing to nf-core/sarek | Exo-seq 相关(建议使用/贡献至 sarek)。 | 16 | – | |
| Human genomics, variants & disease | gwas | UNDER CONSTRUCTION: Genome Wide Association Studies | GWAS(全基因组关联分析)流程(建设中)。 | 27 | – | |
| Human genomics, variants & disease | longraredisease | Long-read sequencing pipeline for rare disease variant discovery | 长读长测序罕见病变异识别流程(神经发育障碍等)。 | 5 | v1.0.0-alpha | |
| Human genomics, variants & disease | mitodetect | A-Z analysis of mitochondrial NGS data | 线粒体 NGS 数据全流程分析。 | 7 | – | |
| Human genomics, variants & disease | oncoanalyser | Comprehensive cancer DNA/RNA analysis and reporting pipeline | 肿瘤 DNA/RNA 综合分析与报告生成流程。 | 97 | 2.3.0 | |
| Human genomics, variants & disease | pacvar | Long-read PacBio sequencing processing for WGS and PureTarget | PacBio 长读长 WGS/PureTarget 测序数据处理流程。 | 13 | 1.0.1 | |
| Human genomics, variants & disease | phaseimpute | Phase and impute genetic data | 遗传数据分相与基因型填补流程。 | 27 | 1.1.0 | |
| Human genomics, variants & disease | radseq | Variant-calling pipeline for RADseq | RADseq 变异检测流程。 | 7 | – | |
| Human genomics, variants & disease | raredisease | Call and score variants from WGS/WES of rare disease patients | 罕见病 WGS/WES 变异检测与打分流程。 | 112 | 2.6.0 | |
| Human genomics, variants & disease | rarevariantburden | Summary count based rare variant burden test (e.g., vs gnomAD) | 基于汇总计数的稀有变异负担检验(可与 gnomAD 等对照)。 | 0 | – | |
| Human genomics, variants & disease | rnadnavar | Integrated RNA+DNA somatic mutation detection | RNA+DNA 联合分析的体细胞突变检测流程。 | 14 | – | |
| Human genomics, variants & disease | sarek | Germline/somatic variant calling + annotation from WGS/targeted | WGS/靶向测序的生殖系/体细胞变异检测与注释(含预处理、calling、annotation)。 | 532 | 3.7.1 | |
| Human genomics, variants & disease | ssds | Single-stranded DNA Sequencing (SSDS) pipeline | SSDS(单链 DNA 测序)分析流程。 | 1 | – | |
| Human genomics, variants & disease | tumourevo | Model tumour clonal evolution from WGS (CN, subclones, signatures) | 基于 WGS 的肿瘤克隆进化建模(CN、亚克隆、突变签名等)。 | 20 | – | |
| Human genomics, variants & disease | variantbenchmarking | Evaluate/validate variant calling accuracy | 变异检测方法准确性评估与验证流程(benchmark)。 | 37 | 1.4.0 | |
| Human genomics, variants & disease | variantcatalogue | Generate population variant catalogues from WGS | 从 WGS 构建人群变异目录(变异列表及频率)。 | 13 | – | |
| Human genomics, variants & disease | variantprioritization | (no description shown) | 变异优先级筛选流程(原表未给出描述)。 | 12 | – | |
| Viruses & pathogen surveillance | pathogensurveillance | Surveillance of pathogens using population genomics and sequencing | 基于群体基因组与测序的病原体监测流程。 | 52 | 1.0.0 | |
| Viruses & pathogen surveillance | phageannotator | Identify, annotate, quantify phage sequences in (meta)genomes | 在(宏)基因组中识别、注释并定量噬菌体序列。 | 17 | – | |
| Viruses & pathogen surveillance | tbanalyzer | Pipeline for Mycobacterium tuberculosis complex analysis | 结核分枝杆菌复合群(MTBC)分析流程。 | 13 | – | |
| Viruses & pathogen surveillance | viralmetagenome | Untargeted viral genome reconstruction with iSNV detection from metagenomes | 宏基因组中无靶向病毒全基因组重建,并检测 iSNV。 | 28 | 1.0.1 | |
| Viruses & pathogen surveillance | viralintegration | Identify viral integration events using chimeric reads | 基于嵌合 reads 的病毒整合事件检测流程。 | 17 | 0.1.1 | |
| Viruses & pathogen surveillance | viralrecon | Viral assembly and intrahost/low-frequency variant calling | 病毒组装与宿主体内/低频变异检测流程。 | 151 | 3.0.0 | |
| Viruses & pathogen surveillance | vipr | Viral assembly and intrahost/low-frequency variant calling | 病毒组装与体内/低频变异检测流程(类似 viralrecon)。 | 14 | – | |
| Metagenomics & microbiome | ampliseq | Amplicon sequencing workflow using DADA2 and QIIME2 | 扩增子测序(如 16S/ITS)分析:DADA2 + QIIME2。 | 231 | 2.15.0 | |
| Metagenomics & microbiome | coproid | Coprolite host identification pipeline | 粪化石(coprolite)宿主鉴定流程。 | 13 | 2.0.0 | |
| Metagenomics & microbiome | createtaxdb | Automated construction of classifier databases for multiple tools | 自动化并行构建多种宏基因组分类工具的数据库。 | 20 | 2.0.0 | |
| Metagenomics & microbiome | detaxizer | Identify (and optionally remove) sequences; default remove human | 识别并(可选)去除特定序列(默认去除人源污染)。 | 22 | 1.3.0 | |
| Metagenomics & microbiome | funcscan | (Meta-)genome screening for functional and natural product genes | (宏)基因组功能基因与天然产物基因簇筛查。 | 99 | 3.0.0 | |
| Metagenomics & microbiome | mag | Assembly and binning of metagenomes | 宏基因组组装与分箱(MAG 构建)。 | 264 | 5.3.0 | |
| Metagenomics & microbiome | magmap | Mapping reads to large collections of genomes | 将 reads 比对到大型基因组集合的最佳实践流程。 | 10 | 1.0.0 | |
| Metagenomics & microbiome | metapep | From metagenomes to epitopes and beyond | 从宏基因组到表位(epitope)等免疫相关下游分析。 | 12 | 1.0.0 | |
| Metagenomics & microbiome | metatdenovo | De novo assembly/annotation of metatranscriptomic or metagenomic data | 宏转录组/宏基因组的从头组装与注释(支持原核/真核/病毒)。 | 34 | 1.3.0 | |
| Metagenomics & microbiome | taxprofiler | Multi-taxonomic profiling of shotgun short/long read metagenomics | shotgun 宏基因组多类群(多生物界)分类谱分析(短读长/长读长)。 | 175 | 1.2.5 | |
| Genome assembly, annotation & comparative genomics | bacass | Simple bacterial assembly and annotation pipeline | 简单的细菌组装与注释流程。 | 80 | 2.5.0 | |
| Genome assembly, annotation & comparative genomics | bactmap | Mapping-based pipeline for bacterial phylogeny from WGS | 基于比对的细菌 WGS 系统发育/建树流程。 | 61 | 1.0.0 | |
| Genome assembly, annotation & comparative genomics | denovohybrid | Hybrid genome assembly pipeline (under construction) | 混合组装流程(长+短读长)(建设中)。 | 8 | – | |
| Genome assembly, annotation & comparative genomics | genomeannotator | Identify (coding) gene structures in draft genomes | 草图基因组(draft genome)基因结构(编码基因)注释流程。 | 34 | – | |
| Genome assembly, annotation & comparative genomics | genomeassembler | Assembly and scaffolding from long ONT/PacBio HiFi reads | 长读长(ONT/PacBio HiFi)基因组组装与脚手架构建。 | 31 | 1.1.0 | |
| Genome assembly, annotation & comparative genomics | genomeqc | Compare quality of multiple genomes and annotations | 比较多个基因组及其注释质量。 | 19 | – | |
| Genome assembly, annotation & comparative genomics | genomeskim | QC/filter genome skims; organelle assembly and/or analysis | genome skim 数据质控/过滤,并进行细胞器组装或相关分析。 | 3 | – | |
| Genome assembly, annotation & comparative genomics | hgtseq | Investigate horizontal gene transfer from NGS data | 从 NGS 数据研究水平基因转移(HGT)。 | 26 | 1.1.0 | |
| Genome assembly, annotation & comparative genomics | multiplesequencealign | Systematically evaluate MSA methods | 多序列比对(MSA)方法系统评估流程。 | 40 | 1.1.1 | |
| Genome assembly, annotation & comparative genomics | neutronstar | De novo assembly for 10x linked-reads using Supernova | 10x linked-reads 从头组装流程(Supernova)。 | 3 | 1.0.0 | |
| Genome assembly, annotation & comparative genomics | pangenome | Render sequences into a pangenome graph | 将序列集合渲染为泛基因组图(pangenome graph)。 | 102 | 1.1.3 | |
| Genome assembly, annotation & comparative genomics | pairgenomealign | Pairwise genome comparison with LAST + plots | 基于 LAST 的两两基因组比对与可视化绘图。 | 10 | 2.2.1 | |
| Genome assembly, annotation & comparative genomics | phyloplace | Phylogenetic placement with EPA-NG | 使用 EPA-NG 的系统发育定位(placement)流程。 | 13 | 2.0.0 | |
| Genome assembly, annotation & comparative genomics | reportho | Comparative analysis of ortholog predictions | 直系同源(ortholog)预测结果的比较分析流程。 | 11 | 1.1.0 | |
| Immunology & antigen presentation | airrflow | AIRR-seq repertoire analysis using Immcantation | 免疫受体库(BCR/TCR,AIRR-seq)分析:基于 Immcantation。 | 73 | 4.3.1 | |
| Immunology & antigen presentation | epitopeprediction | Epitope prediction and annotation pipeline | 表位(epitope)预测与注释流程。 | 50 | 3.1.0 | |
| Immunology & antigen presentation | hlatyping | Precision HLA typing from NGS data | 基于 NGS 的高精度 HLA 分型流程。 | 76 | 2.1.0 | |
| Immunology & antigen presentation | mhcquant | Identify and quantify MHC eluted peptides from MS raw data | 从质谱原始数据识别并定量 MHC 洗脱肽段。 | 42 | 3.1.0 | |
| Proteomics, metabolomics & protein informatics | ddamsproteomics | Quantitative shotgun MS proteomics | 定量 shotgun 质谱蛋白组流程。 | 4 | – | |
| Proteomics, metabolomics & protein informatics | diaproteomics | Automated quantitative analysis of DIA proteomics MS measurements | DIA 蛋白组质谱数据自动化定量分析流程。 | 21 | 1.2.4 | |
| Proteomics, metabolomics & protein informatics | kmermaid | k-mer similarity analysis pipeline | k-mer 相似性分析流程。 | 23 | 0.1.0-alpha | |
| Proteomics, metabolomics & protein informatics | metaboigniter | Metabolomics MS pre-processing with identification/quantification (MS1/MS2) | 代谢组质谱预处理:基于 MS1/MS2 的鉴定与定量。 | 24 | 2.0.1 | |
| Proteomics, metabolomics & protein informatics | proteinannotator | Protein fasta → annotations | 蛋白序列(FASTA)到注释的自动化流程。 | 8 | – | |
| Proteomics, metabolomics & protein informatics | proteinfamilies | Generation and updating of protein families | 蛋白家族的生成与更新流程。 | 21 | 2.2.0 | |
| Proteomics, metabolomics & protein informatics | proteinfold | Protein 3D structure prediction pipeline | 蛋白三维结构预测流程。 | 94 | 1.1.1 | |
| Proteomics, metabolomics & protein informatics | proteogenomicsdb | Generate protein databases for proteogenomics analysis | 构建蛋白基因组学分析所需的蛋白数据库。 | 7 | 1.0.0 | |
| Proteomics, metabolomics & protein informatics | proteomicslfq | Proteomics label-free quantification (LFQ) analysis pipeline | 蛋白组无标记定量(LFQ)分析流程。 | 37 | 1.0.0 | |
| Proteomics, metabolomics & protein informatics | quantms | Quantitative MS workflow (DDA-LFQ, DDA-Isobaric, DIA-LFQ) | 定量蛋白组流程:支持 DDA-LFQ、等标记 DDA、DIA-LFQ 等。 | 34 | 1.2.0 | |
| Proteomics, metabolomics & protein informatics | ribomsqc | QC pipeline monitoring MS performance in ribonucleoside analysis | 核苷相关质谱分析的性能监控与质控流程。 | 0 | – | |
| Imaging & other modalities | cellpainting | (no description shown) | Cell Painting 相关流程(原表未给出描述)。 | 8 | – | |
| Imaging & other modalities | imcyto | Image Mass Cytometry analysis pipeline | 成像质谱细胞术(IMC)图像/数据分析流程。 | 26 | 1.0.0 | |
| Imaging & other modalities | liverctanalysis | UNDER CONSTRUCTION: pipeline for liver CT analysis | 肝脏 CT 影像分析流程(建设中)。 | 0 | – | |
| Imaging & other modalities | lsmquant | Process and analyze light-sheet microscopy images | 光片显微(light-sheet)图像处理与分析流程。 | 5 | – | |
| Imaging & other modalities | mcmicro | Whole-slide multi-channel image processing to single-cell data | 多通道全切片图像到单细胞数据的端到端处理流程。 | 29 | – | |
| Imaging & other modalities | rangeland | Remotely sensed imagery pipeline for land-cover trend files | 遥感影像处理流程:结合辅助数据生成土地覆盖变化趋势文件。 | 9 | 1.0.0 | |
| Imaging & other modalities | troughgraph | Quantitative assessment of permafrost landscapes and thaw level | 冻土景观与冻融程度的定量评估流程。 | 2 | – | |
| Data acquisition, QC & utilities | bamtofastq | Convert BAM/CRAM to FASTQ and perform QC | BAM/CRAM 转 FASTQ 并进行质控。 | 31 | 2.2.0 | |
| Data acquisition, QC & utilities | datasync | System operation / automation workflows | 系统运维/自动化工作流(数据同步与操作任务)。 | 10 | – | |
| Data acquisition, QC & utilities | demo | Simple nf-core style pipeline for workshops and demos | nf-core 风格的示例/教学演示流程。 | 10 | 1.0.2 | |
| Data acquisition, QC & utilities | demultiplex | Demultiplexing pipeline for sequencing data | 测序数据拆样/解复用流程。 | 52 | 1.7.0 | |
| Data acquisition, QC & utilities | fastqrepair | Recover corrupted FASTQ.gz, fix reads, remove unpaired, reorder | 修复损坏 FASTQ.gz:修正不合规 reads、移除未配对 reads、重排序等。 | 6 | 1.0.0 | |
| Data acquisition, QC & utilities | fastquorum | Produce consensus reads using UMIs/barcodes | 基于 UMI/条形码生成共识 reads 的流程。 | 27 | 1.2.0 | |
| Data acquisition, QC & utilities | fetchngs | Fetch metadata and raw FastQ files from public databases | 从公共数据库抓取元数据与原始 FASTQ。 | 185 | 1.12.0 | |
| Data acquisition, QC & utilities | nanoseq | Nanopore demultiplexing, QC and alignment pipeline | Nanopore 数据拆样、质控与比对流程。 | 218 | 3.1.0 | |
| Data acquisition, QC & utilities | readsimulator | Simulate sequencing reads (amplicon, metagenome, WGS, etc.) | 测序 reads 模拟流程(扩增子、靶向捕获、宏基因组、全基因组等)。 | 33 | 1.0.1 | |
| Data acquisition, QC & utilities | references | Build references for multiple use cases | 多用途参考资源构建流程。 | 19 | 0.1 | |
| Data acquisition, QC & utilities | seqinspector | QC-only pipeline producing global/group-specific MultiQC reports | 纯质控流程:运行多种 QC 工具并输出全局/分组 MultiQC 报告。 | 16 | – | |
| Data acquisition, QC & utilities | seqsubmit | Submit data to ENA | 向 ENA 提交数据的流程。 | 3 | – | |
| Genome editing & screens | crisprseq | CRISPR edited data analysis (targeted + screens) | CRISPR 编辑数据分析:靶向编辑质量评估与 pooled screen 关键基因发现。 | 53 | 2.3.0 | |
| Genome editing & screens | crisprvar | Evaluate outcomes from genome editing experiments (WIP) | 基因编辑实验结果评估流程(WIP)。 | 5 | – | |
| Other methods / modelling / non-bio | deepmodeloptim | Stochastic Testing and Input Manipulation for Unbiased Learning Systems | 无偏学习系统的随机测试与输入操控(机器学习相关)。 | 28 | – | |
| Other methods / modelling / non-bio | deepmutscan | Deep mutational scanning (DMS) analysis pipeline | 深度突变扫描(DMS)数据分析流程。 | 3 | – | |
| Other methods / modelling / non-bio | diseasemodulediscovery | Network-based disease module identification | 基于网络的疾病模块识别流程。 | 5 | – | |
| Other methods / modelling / non-bio | drugresponseeval | Evaluate drug response prediction models | 药物反应预测模型的评估流程(统计与生物学上更严谨)。 | 24 | 1.1.0 | |
| Other methods / modelling / non-bio | meerpipe | Astronomy pipeline for MeerKAT pulsar data | MeerKAT 脉冲星数据天文处理流程(成像与计时分析)。 | 10 | – | |
| Other methods / modelling / non-bio | omicsgenetraitassociation | Multi-omics integration and trait association analysis pipeline | 多组学整合并进行性状/表型关联分析的流程。 | 11 | – | |
| Other methods / modelling / non-bio | spinningjenny | Simulating the first industrial revolution using agent-based models | 基于主体(Agent-based)模型模拟第一次工业革命的流程。 | 4 | – |
author: “”
date: ‘r format(Sys.time(), "%d %m %Y")‘
header-includes:
#install.packages(c("picante", "rmdformats"))
#mamba install -c conda-forge freetype libpng harfbuzz fribidi
#mamba install -c conda-forge r-systemfonts r-svglite r-kableExtra freetype fontconfig harfbuzz fribidi libpng
library(knitr)
library(rmdformats)
library(readxl)
library(dplyr)
library(kableExtra)
library(openxlsx)
library(DESeq2)
library(writexl)
options(max.print="75")
knitr::opts_chunk$set(fig.width=8,
fig.height=6,
eval=TRUE,
cache=TRUE,
echo=TRUE,
prompt=FALSE,
tidy=FALSE,
comment=NA,
message=FALSE,
warning=FALSE)
opts_knit$set(width=85)
#rmarkdown::render('Phyloseq_Group9_10_11_pre-FMT.Rmd',output_file='Phyloseq_Group9_10_11_pre-FMT.html')
# Phyloseq R library
#* Phyloseq web site : https://joey711.github.io/phyloseq/index.html
#* See in particular tutorials for
# - importing data: https://joey711.github.io/phyloseq/import-data.html
# - heat maps: https://joey711.github.io/phyloseq/plot_heatmap-examples.htmlImport raw data and assign sample key:
#extend qiime2_metadata_for_qza_to_phyloseq.tsv with Diet and Flora
#setwd("~/DATA/Data_Laura_16S_2/core_diversity_e4753")
#map_corrected <- read.csv("qiime2_metadata_for_qza_to_phyloseq.tsv", sep="\t", row.names=1)
#knitr::kable(map_corrected) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))install.packages("dplyr") # To manipulate dataframes
install.packages("readxl") # To read Excel files into R
install.packages("ggplot2") # for high quality graphics
install.packages("heatmaply")
source("https://bioconductor.org/biocLite.R")
biocLite("phyloseq")#mamba install -c conda-forge r-ggplot2 r-vegan r-data.table
#BiocManager::install("microbiome")
#install.packages("ggpubr")
#install.packages("heatmaply")
library("readxl") # necessary to import the data from Excel file
library("ggplot2") # graphics
library("picante")
library("microbiome") # data analysis and visualisation
library("phyloseq") # also the basis of data object. Data analysis and visualisation
library("ggpubr") # publication quality figures, based on ggplot2
library("dplyr") # data handling, filter and reformat data frames
library("RColorBrewer") # nice color options
library("heatmaply")
library(vegan)
library(gplots)
#install.packages("openxlsx")
library(openxlsx)Three tables are needed
library(tidyr)
# For QIIME1
#ps.ng.tax <- import_biom("./exported_table/feature-table.biom", "./exported-tree/tree.nwk")
# For QIIME2
#install.packages("remotes")
#remotes::install_github("jbisanz/qiime2R")
#"core_metrics_results/rarefied_table.qza", rarefying performed in the code, therefore import the raw table.
library(qiime2R)
ps_raw <- qza_to_phyloseq(
features = "table.qza", #cp ../Data_Karoline_16S_2025/dada2_tests2/test_7_f240_r240/table.qza .
tree = "rooted-tree.qza", #cp ../Data_Karoline_16S_2025/rooted-tree.qza .
metadata = "qiime2_metadata_for_qza_to_phyloseq.tsv" #cp ../Data_Karoline_16S_2025/qiime2_metadata_for_qza_to_phyloseq.tsv .
)
# or
#biom convert \
# -i ./exported_table/feature-table.biom \
# -o ./exported_table/feature-table-v1.biom \
# --to-json
#ps_raw <- import_biom("./exported_table/feature-table-v1.biom", treefilename="./exported-tree/tree.nwk")
sample <- read.csv("./qiime2_metadata_for_qza_to_phyloseq.tsv", sep="\t", row.names=1)
SAM = sample_data(sample, errorIfNULL = T)
#> setdiff(rownames(SAM), sample_names(ps_raw))
#[1] "sample-L9" should be removed since the low reads
ps_base <- merge_phyloseq(ps_raw, SAM)
print(ps_base)
taxonomy <- read.delim("taxonomy.tsv", sep="\t", header=TRUE) #cp ../Data_Karoline_16S_2025/exported-taxonomy/taxonomy.tsv .
#head(taxonomy)
# Separate taxonomy string into separate ranks
taxonomy_df <- taxonomy %>% separate(Taxon, into = c("Domain","Phylum","Class","Order","Family","Genus","Species"), sep = ";", fill = "right", extra = "drop")
# Use Feature.ID as rownames
rownames(taxonomy_df) <- taxonomy_df$Feature.ID
taxonomy_df <- taxonomy_df[, -c(1, ncol(taxonomy_df))] # Drop Feature.ID and Confidence
# Create tax_table
tax_table_final <- phyloseq::tax_table(as.matrix(taxonomy_df))
# Merge tax_table with existing phyloseq object
ps_base <- merge_phyloseq(ps_base, tax_table_final)
# Check
ps_base
#colnames(phyloseq::tax_table(ps_base)) <- c("Domain","Phylum","Class","Order","Family","Genus","Species")
saveRDS(ps_base, "./ps_base.rds")Visualize data
sample_names(ps_base)
rank_names(ps_base)
sample_variables(ps_base)
# Define sample names once
samples <- c(
#"sample-A1","sample-A2","sample-A5","sample-A6","sample-A7","sample-A8","sample-A9","sample-A10", #RESIZED: "sample-A3","sample-A4","sample-A11",
#"sample-B1","sample-B2","sample-B3","sample-B4","sample-B5","sample-B6","sample-B7", #RESIZED: "sample-B8","sample-B9","sample-B10","sample-B11","sample-B12","sample-B13","sample-B14","sample-B15","sample-B16",
#"sample-C1","sample-C2","sample-C3","sample-C4","sample-C5","sample-C6","sample-C7", #RESIZED: "sample-C8","sample-C9","sample-C10",
#"sample-E1","sample-E2","sample-E3","sample-E4","sample-E5","sample-E6","sample-E7","sample-E8","sample-E9","sample-E10", #RESIZED:
#"sample-F1","sample-F2","sample-F3","sample-F4","sample-F5",
"sample-G1","sample-G2","sample-G3","sample-G4","sample-G5","sample-G6",
"sample-H1","sample-H2","sample-H3","sample-H4","sample-H5","sample-H6",
"sample-I1","sample-I2","sample-I3","sample-I4","sample-I5","sample-I6",
"sample-J1","sample-J2","sample-J3","sample-J4","sample-J10","sample-J11", #RESIZED: "sample-J5","sample-J8","sample-J9", "sample-J6","sample-J7",
"sample-K1","sample-K2","sample-K3","sample-K4","sample-K5","sample-K6", #RESIZED: "sample-K7","sample-K8","sample-K9","sample-K10", "sample-K11","sample-K12","sample-K13","sample-K14","sample-K15",
"sample-L2","sample-L3","sample-L4","sample-L5","sample-L6" #RESIZED:"sample-L1","sample-L7","sample-L8","sample-L10","sample-L11","sample-L12","sample-L13","sample-L14","sample-L15",
#"sample-M1","sample-M2","sample-M3","sample-M4","sample-M5","sample-M6","sample-M7","sample-M8",
#"sample-N1","sample-N2","sample-N3","sample-N4","sample-N5","sample-N6","sample-N7","sample-N8","sample-N9","sample-N10",
#"sample-O1","sample-O2","sample-O3","sample-O4","sample-O5","sample-O6","sample-O7","sample-O8"
)
ps_pruned <- prune_samples(samples, ps_base)
sample_names(ps_pruned)
rank_names(ps_pruned)
sample_variables(ps_pruned)No samples were excluded as low-depth outliers (library sizes below the minimum depth threshold of 1,000 reads), and the remaining dataset (ps_filt) contains only samples meeting this depth cutoff with taxa retained only if they have nonzero total counts.
# ------------------------------------------------------------
# Filter low-depth samples (recommended for all analyses)
# ------------------------------------------------------------
min_depth <- 1000 # <-- adjust to your data / study design, keeps all!
ps_filt <- prune_samples(sample_sums(ps_pruned) >= min_depth, ps_pruned)
ps_filt <- prune_taxa(taxa_sums(ps_filt) > 0, ps_filt)
# Keep a depth summary for reporting / QC
depth_summary <- summary(sample_sums(ps_filt))
depth_summaryDifferential abundance (DESeq2) → ps_deseq: non-rarefied integer counts derived from ps_filt, with optional count-based taxon prefilter
(default: taxa total counts ≥ 10 across all samples)
From ps_filt (e.g. 5669 taxa and 239 samples), we branch into analysis-ready objects in two directions:
Direction 1 for diversity analyses
ps_rarefied ✅ (common)ps_rarefied ✅ (often recommended)ps_rel or Hellinger ✅ (rarefaction optional)Normalize number of reads in each sample using median sequencing depth.
# RAREFACTION
set.seed(9242) # This will help in reproducing the filtering and nomalisation.
ps_rarefied <- rarefy_even_depth(ps_filt, sample.size = 6389)
#total <- 6389
# # NORMALIZE number of reads in each sample using median sequencing depth.
# total = median(sample_sums(ps.ng.tax))
# #> total
# #[1] 42369
# standf = function(x, t=total) round(t * (x / sum(x)))
# ps.ng.tax = transform_sample_counts(ps.ng.tax, standf)
# ps_rel <- microbiome::transform(ps.ng.tax, "compositional")
#
# saveRDS(ps.ng.tax, "./ps.ng.tax.rds")Direction 2 for taxonomic composition plots
ps_rel: relative abundance (compositional) computed after sample filtering (e.g. 5669 taxa and 239 samples)ps_abund / ps_abund_rel: taxa filtered for plotting (e.g., keep taxa with mean relative abundance > 0.1%); (e.g. 95 taxa and 239 samples)
ps_abund = counts, ps_abund_rel = relative abundance (use for visualization, not DESeq2)For the heatmaps, we focus on the most abundant OTUs by first converting counts to relative abundances within each sample. We then filter to retain only OTUs whose mean relative abundance across all samples exceeds 0.1% (0.001). We are left with 199 OTUs which makes the reading much more easy.
# 1) Convert to relative abundances
ps_rel <- transform_sample_counts(ps_filt, function(x) x / sum(x))
# 2) Get the logical vector of which OTUs to keep (based on relative abundance)
keep_vector <- phyloseq::filter_taxa(
ps_rel,
function(x) mean(x) > 0.001,
prune = FALSE
)
# 3) Use the TRUE/FALSE vector to subset absolute abundance data
ps_abund <- prune_taxa(names(keep_vector)[keep_vector], ps_filt)
# 4) Normalize the final subset to relative abundances per sample
ps_abund_rel <- transform_sample_counts(
ps_abund,
function(x) x / sum(x)
) library(stringr)
#for id in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206; do
#for id in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62; do
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Domain\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Domain\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Phylum\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Phylum\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Class\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Class\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Order\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Order\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Family\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Family\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Genus\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Genus\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Species\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Species\"], \"__\")[[1]][2]"
#done
phyloseq::tax_table(ps_abund_rel)[1,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Species"], "__")[[1]][2]
#aes(color="Phylum", fill="Phylum") --> aes()
#ggplot(data=data, aes(x=Sample, y=Abundance, fill=Phylum))
#options(max.print = 1e6)
my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, fill="Phylum") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=2)) #6 instead of theme.size\pagebreak Regroup together pre vs post stroke samples and normalize number of reads in each group using median sequencing depth.
ps_abund_rel_group <- merge_samples(ps_abund_rel, "G9_10_11_preFMT")
#PENDING: The effect weighted twice by sum(x), is the same to the effect weighted once directly from absolute abundance?!
ps_abund_rel_group_ = transform_sample_counts(ps_abund_rel_group, function(x) x / sum(x))
#plot_bar(ps_abund_relSampleType_, fill = "Phylum") + geom_bar(aes(color=Phylum, fill=Phylum), stat="identity", position="stack")
plot_bar(ps_abund_rel_group_, fill="Phylum") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
) my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, fill="Class") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=3))Regroup together pre vs post stroke samples and normalize number of reads in each group using median sequencing depth.
plot_bar(ps_abund_rel_group_, fill="Class") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)\pagebreak
my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, fill="Order") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=4))Regroup together pre vs post stroke and normalize number of reads in each group using median sequencing depth.
plot_bar(ps_abund_rel_group_, fill="Order") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)\pagebreak
my_colors <- c(
"#FF0000", "#000000", "#0000FF", "#C0C0C0", "#FFFFFF", "#FFFF00", "#00FFFF", "#FFA500", "#00FF00", "#808080", "#FF00FF", "#800080", "#FDD017", "#0000A0", "#3BB9FF", "#008000", "#800000", "#ADD8E6", "#F778A1", "#800517", "#736F6E", "#F52887", "#C11B17", "#5CB3FF", "#A52A2A", "#FF8040", "#2B60DE", "#736AFF", "#1589FF", "#98AFC7", "#8D38C9", "#307D7E", "#F6358A", "#151B54", "#6D7B8D", "#FDEEF4", "#FF0080", "#F88017", "#2554C7", "#FFF8C6", "#D4A017", "#306EFF", "#151B8D", "#9E7BFF", "#EAC117", "#E0FFFF", "#15317E", "#6C2DC7", "#FBB917", "#FCDFFF", "#15317E", "#254117", "#FAAFBE", "#357EC7"
)
plot_bar(ps_abund_rel, fill="Family") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=8))Regroup together pre vs post stroke samples and normalize number of reads in each group using median sequencing depth.
plot_bar(ps_abund_rel_group_, fill="Family") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)\pagebreak
\pagebreak
Plot Chao1 richness estimator, Observed OTUs, Shannon index, and Phylogenetic diversity. Regroup together samples from the same group.
# using rarefied data
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even42369.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering/rep_set.tre
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even4753.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering_stool/rep_set.tre
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even4753.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering_swab/rep_set.tre
library(dplyr)
library(reshape2)
library(ggpubr)
library(phyloseq)
library(kableExtra)
# ------------------------------------------------------------
# 0) Restrict analysis to the specified sample subset
# ------------------------------------------------------------
stopifnot(exists("samples"))
stopifnot(exists("ps_rarefied"))
ps_rarefied_sub <- prune_samples(samples, ps_rarefied)
hmp.meta <- meta(ps_rarefied_sub)
hmp.meta$sam_name <- rownames(hmp.meta)
# ------------------------------------------------------------
# 1) Read QIIME2 alpha-diversity outputs
# ------------------------------------------------------------
shannon <- read.table("exported_alpha/shannon/alpha-diversity.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
faith_pd <- read.table("exported_alpha/faith_pd/alpha-diversity.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
observed <- read.table("exported_alpha/observed_features/alpha-diversity.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
colnames(shannon) <- c("sam_name", "shannon")
colnames(faith_pd) <- c("sam_name", "PD_whole_tree")
colnames(observed) <- c("sam_name", "observed_otus")
div.df <- Reduce(function(x, y) merge(x, y, by = "sam_name", all = FALSE),
list(shannon, faith_pd, observed))
# ------------------------------------------------------------
# 2) Merge in metadata (from the pruned phyloseq object)
# ------------------------------------------------------------
div.df <- merge(div.df, hmp.meta, by = "sam_name", all.x = TRUE, all.y = FALSE)
# Keep ONLY samples from the predefined list (safety net)
div.df <- div.df %>% filter(sam_name %in% samples)
# ------------------------------------------------------------
# 3) Collapse groups: Group6/7/8 -> pre-FMT; keep only 4 groups
# ------------------------------------------------------------
div.df <- div.df %>%
mutate(
Group4 = case_when(
Group %in% c("Group6","Group7","Group8") ~ "pre-FMT",
Group %in% c("Group9","Group10","Group11") ~ as.character(Group),
TRUE ~ NA_character_
),
Group4 = factor(Group4, levels = c("pre-FMT","Group9","Group10","Group11"))
) %>%
filter(!is.na(Group4))
# ------------------------------------------------------------
# 4) Reformat table for reporting
# ------------------------------------------------------------
div.df2 <- div.df[, c("sam_name", "Group4", "shannon", "observed_otus", "PD_whole_tree")]
colnames(div.df2) <- c("Sample name", "Group", "Shannon", "OTU", "Phylogenetic Diversity")
write.csv(div.df2, file = "alpha_diversities_G9_10_11_preFMT.csv", row.names = FALSE)
#knitr::kable(div.df2) %>%
# kable_styling(bootstrap_options = c("striped","hover","condensed","responsive"))
# ------------------------------------------------------------
# 5) QC: print which samples were used
# ------------------------------------------------------------
requested_samples <- samples
present_in_ps <- sample_names(ps_rarefied)
missing_in_ps <- setdiff(requested_samples, present_in_ps)
cat("\n==== QC: Sample selection (ps_rarefied) ====\n")
cat("Requested samples:", length(requested_samples), "\n")
cat("Present in ps_rarefied:", length(present_in_ps), "\n")
cat("Missing from ps_rarefied:", length(missing_in_ps), "\n")
if (length(missing_in_ps) > 0) print(missing_in_ps)
used_samples <- div.df2$`Sample name`
missing_in_alpha <- setdiff(requested_samples, used_samples)
extra_in_alpha <- setdiff(used_samples, requested_samples)
cat("\n==== QC: Samples used in alpha-div df (div.df2) ====\n")
cat("Used samples:", length(used_samples), "\n")
cat("Requested but NOT used:", length(missing_in_alpha), "\n")
if (length(missing_in_alpha) > 0) print(missing_in_alpha)
cat("Used but NOT requested (should be 0):", length(extra_in_alpha), "\n")
if (length(extra_in_alpha) > 0) print(extra_in_alpha)
qc_table <- div.df %>%
select(sam_name, Group, Group4) %>%
distinct() %>%
arrange(Group4, Group, sam_name)
#cat("\n==== QC: Sample -> Group mapping used for plotting ====\n")
#print(qc_table)
#
#cat("\n==== QC: Counts per collapsed group (Group4) ====\n")
#print(div.df$Group4)
# ------------------------------------------------------------
# 6) Melt + plot
# ------------------------------------------------------------
div_df_melt <- reshape2::melt(
div.df2,
id.vars = c("Sample name","Group"),
variable.name = "variable",
value.name = "value"
)
# ensure final group order
div_df_melt$Group <- factor(div_df_melt$Group, levels = c("pre-FMT","Group9","Group10","Group11"))
#p <- ggboxplot(div_df_melt, x = "Group", y = "value",
# facet.by = "variable",
# scales = "free",
# width = 0.5,
# fill = "gray", legend = "right") +
# theme(
# axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1, size = 8),
# axis.text.y = element_text(size = 10),
# axis.title.x = element_text(size = 12),
# axis.title.y = element_text(size = 12)
# )
# all pairwise comparisons among the 4 groups
#lev <- levels(droplevels(div_df_melt$Group))
#L.pairs <- combn(lev, 2, simplify = FALSE)
#
#p2 <- p + stat_compare_means(
# method = "wilcox.test",
# comparisons = L.pairs,
# label = "p.signif",
# p.adjust.method = "BH",
# hide.ns = FALSE,
# symnum.args = list(
# cutpoints = c(0, 0.0001, 0.001, 0.01, 0.05, 1),
# symbols = c("****", "***", "**", "*", "ns")
# )
#)
#p2
# ------------------------------------------------------------
# 7) Save figure
# ------------------------------------------------------------
#ggsave("./figures/alpha_diversity_G9_10_11_preFMT.png", p2, device="png", height = 10, width = 15, dpi = 300)
#ggsave("./figures/alpha_diversity_G9_10_11_preFMT.svg", p2, device="svg", height = 10, width = 15)
# ------------------------------------------------------------
# 8) Save statistics
# ------------------------------------------------------------
library(dplyr)
library(purrr)
# same pair list as in the plot
lev <- levels(droplevels(div_df_melt$Group))
L.pairs <- combn(lev, 2, simplify = FALSE)
# Pairwise Wilcoxon per metric, with BH adjustment (matches p.adjust.method="BH")
stats_for_plot <- div_df_melt %>%
group_by(variable) %>%
group_modify(~{
dat <- .x
res <- purrr::map_dfr(L.pairs, function(pr) {
g1 <- pr[1]; g2 <- pr[2]
x1 <- dat$value[dat$Group == g1]
x2 <- dat$value[dat$Group == g2]
wt <- suppressWarnings(wilcox.test(x1, x2, exact = FALSE))
tibble(
group1 = g1,
group2 = g2,
n1 = length(x1),
n2 = length(x2),
p = unname(wt$p.value)
)
})
res %>%
mutate(
p.adj = p.adjust(p, method = "BH"),
p.signif = case_when(
p.adj <= 0.0001 ~ "****",
p.adj <= 0.001 ~ "***",
p.adj <= 0.01 ~ "**",
p.adj <= 0.05 ~ "*",
TRUE ~ "ns"
)
)
}) %>%
ungroup() %>%
rename(Metric = variable) %>%
arrange(Metric, group1, group2)
# Print the exact statistics used for annotation
print(stats_for_plot, n = Inf)
write.csv(stats_for_plot, "./figures/alpha_diversity_pairwise_stats_used_for_plot.csv", row.names = FALSE)
library(openxlsx)
# (optional) make sure the folder exists
dir.create("./figures", showWarnings = FALSE, recursive = TRUE)
out_xlsx <- "./figures/alpha_diversity_pairwise_stats_used_for_plot.xlsx"
write.xlsx(
x = list("pairwise_stats" = stats_for_plot),
file = out_xlsx,
overwrite = TRUE
)
cat("Saved Excel file to: ", out_xlsx, "\n", sep = "")# ---------------------------------------------------------------------------------
# ----------------------------- draw plots from input -----------------------------
library(dplyr)
library(ggpubr)
library(reshape2)
# Example: read from a CSV file you wrote earlier
# stats_for_plot <- read.csv("./figures/alpha_diversity_pairwise_stats_used_for_plot.csv", stringsAsFactors = FALSE)
# Or if you paste as text:
txt <- 'Metric,group1,group2,n1,n2,p,p.adj,p.signif
Shannon,Group10,Group11,6,5,1,1,ns
Shannon,Group9,Group10,6,6,0.297953061608168,0.595906123216336,ns
Shannon,Group9,Group11,6,5,0.522816653919089,0.631819188094748,ns
Shannon,pre-FMT,Group10,18,6,0.017949090591566,0.0538472717746981,ns
Shannon,pre-FMT,Group11,18,5,0.00651735384842052,0.0391041230905231,*
Shannon,pre-FMT,Group9,18,6,0.526515990078957,0.631819188094748,ns
OTU,Group10,Group11,6,5,0.143214612017615,0.214821918026422,ns
OTU,Group9,Group10,6,6,0.229766270461138,0.275719524553366,ns
OTU,Group9,Group11,6,5,0.522816653919089,0.522816653919089,ns
OTU,pre-FMT,Group10,18,6,0.0015274145728676,0.00916448743720559,**
OTU,pre-FMT,Group11,18,5,0.0675601586409843,0.135120317281969,ns
OTU,pre-FMT,Group9,18,6,0.00759484512604444,0.0227845353781333,*
Phylogenetic Diversity,Group10,Group11,6,5,1,1,ns
Phylogenetic Diversity,Group9,Group10,6,6,1,1,ns
Phylogenetic Diversity,Group9,Group11,6,5,0.927264473525232,1,ns
Phylogenetic Diversity,pre-FMT,Group10,18,6,0.000361550896811121,0.00108465269043336,**
Phylogenetic Diversity,pre-FMT,Group11,18,5,0.000910436385913111,0.00182087277182622,**
Phylogenetic Diversity,pre-FMT,Group9,18,6,0.000361550896811121,0.00108465269043336,**'
stats_for_plot <- read.csv(textConnection(txt), stringsAsFactors = FALSE)
# Match naming to your plot data: your facet variable is called "variable"
stats_for_plot <- stats_for_plot %>%
rename(variable = Metric) %>%
mutate(
group1 = factor(group1, levels = c("pre-FMT","Group9","Group10","Group11")),
group2 = factor(group2, levels = c("pre-FMT","Group9","Group10","Group11"))
)
# ------------------------------------------------------------
# Melt + plot (same as yours)
# ------------------------------------------------------------
div_df_melt <- reshape2::melt(
div.df2,
id.vars = c("Sample name","Group"),
variable.name = "variable",
value.name = "value"
)
div_df_melt$Group <- factor(div_df_melt$Group, levels = c("pre-FMT","Group9","Group10","Group11"))
p <- ggboxplot(div_df_melt, x = "Group", y = "value",
facet.by = "variable",
scales = "free",
width = 0.5,
fill = "gray", legend = "right") +
theme(
axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1, size = 8),
axis.text.y = element_text(size = 10),
axis.title.x = element_text(size = 12),
axis.title.y = element_text(size = 12)
)
# ------------------------------------------------------------
# Add significance from your table (NO recomputation)
# ------------------------------------------------------------
# Compute y positions per facet so brackets don't overlap
ymax_by_metric <- div_df_melt %>%
group_by(variable) %>%
summarise(ymax = max(value, na.rm = TRUE), .groups = "drop")
stats_for_plot2 <- stats_for_plot %>%
left_join(ymax_by_metric, by = "variable") %>%
group_by(variable) %>%
arrange(p.adj) %>%
mutate(
# stack brackets within each facet
y.position = ymax * (1.05 + 0.08 * (row_number() - 1)),
# what to display (stars or "ns")
label = p.signif
) %>%
ungroup()
p2 <- p +
stat_pvalue_manual(
stats_for_plot2,
label = "label",
xmin = "group1",
xmax = "group2",
y.position = "y.position",
tip.length = 0.01,
hide.ns = FALSE
)
p2
# ------------------------------------------------------------
# Save
# ------------------------------------------------------------
ggsave("./figures/alpha_diversity_G9_10_11_preFMT_from_table.png",
p2, device="png", height = 6, width = 9, dpi = 300)
ggsave("./figures/alpha_diversity_G9_10_11_preFMT_from_table.svg",
p2, device="svg", height = 6, width = 9)
# label = sprintf("BH p=%.3g", p.adj)
Pairwise PERMANOVA tests were performed on Bray–Curtis distance matrices to compare bacterial community composition between all pairs of sample groups (metadata column Group). For each pairwise comparison, the distance matrix was subset to samples from the two groups only, and significance was assessed using vegan::adonis2 with 9,999 permutations. Resulting p-values were adjusted for multiple testing using both Benjamini–Hochberg (BH/FDR) and Bonferroni corrections.
Bray_pairwise_PERMANOVA <- read.csv("figures_MP_Group9_10_11_PreFMT/Bray_pairwise_PERMANOVA.csv", sep = ",")
knitr::kable(Bray_pairwise_PERMANOVA, caption = "Pairwise PERMANOVA results (distance-based community differences among Group levels).") %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))# --- Global beta-diversity (PERMANOVA) ---
cat("```text\n")
cat(
"[PERMANOVA result]\n",
"The object contained internal attribute: PCoA ADONIS\n",
"Permutation test for adonis under reduced model\n",
"Permutation: free\n",
"Number of permutations: 9999\n",
"\n",
"vegan::adonis2(formula = .formula, data = sampleda, permutations = permutations, method = distmethod)\n",
" Df SumOfSqs R2 F Pr(>F) \n",
"Model 3 5.3256 0.58559 14.602 1e-04 ***\n",
"Residual 31 3.7689 0.41441 \n",
"Total 34 9.0945 1.00000 \n",
"---\n",
"Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1\n",
sep = ""
)
cat("```\n")# --- Global beta-diversity (PERMANOVA) ---
cat("```text\n")
cat(
"[PERMANOVA result]\n",
"The object contained internal attribute: PCoA ADONIS\n",
"Permutation test for adonis under reduced model\n",
"Permutation: free\n",
"Number of permutations: 9999\n",
"\n",
"vegan::adonis2(formula = .formula, data = sampleda, permutations = permutations, method = distmethod)\n",
" Df SumOfSqs R2 F Pr(>F) \n",
"Model 2 0.82208 0.3324 3.4853 1e-04 ***\n",
"Residual 14 1.65109 0.6676 \n",
"Total 16 2.47317 1.0000 \n",
"---\n",
"Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1\n",
sep = ""
)
cat("```\n")knitr::include_graphics("figures_MP_Group9_10_11_PreFMT/PCoA3.png")knitr::include_graphics("figures_MP_Group9_10_11/PCoA3.png")Differential abundance analysis aims to find the differences in the abundance of each taxa between two groups of samples, assigning a significance value to each comparison.
# ------------------------------------------------------------
# DESeq2: non-rarefied integer counts + optional taxon prefilter
# ------------------------------------------------------------
ps_deseq <- ps_filt
ps_deseq_sel2 <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel2) <- otu_table(ps_deseq)[,c("sample-J1","sample-J2","sample-J3","sample-J4","sample-J10","sample-J11", "sample-K1","sample-K2","sample-K3","sample-K4","sample-K5","sample-K6")]
diagdds = phyloseq_to_deseq2(ps_deseq_sel2, ~Group)
diagdds$Group <- relevel(diagdds$Group, "Group9")
diagdds = DESeq(diagdds, test="Wald", fitType="parametric")
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel2)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group9_vs_Group10"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics("DEGs_Group9_vs_Group10.png")Microbiome analysis
Sample collection and storage
Fecal samples were collected from mice between 9–10 am at the indicated timepoints and immediately frozen at −80 °C until use. DNA extraction
Genomic DNA was extracted from fecal pellets using the QIAamp Fast DNA Stool Mini Kit (Qiagen, #51604) following a protocol optimized for murine stool. Briefly, pellets were mechanically homogenized in a Precellys® 24 homogenizer (Bertin Technologies) using Soil grinding tubes (Bertin Technologies, #SK38), and DNA was extracted according to the manufacturer’s instructions. DNA yield was measured with NanoDrop, and the concentration was adjusted to 10 ng/µl. 16S rRNA gene amplicon library preparation and sequencing
16S rRNA amplicons (V3–V4 region) were generated using degenerate primers that contain Illumina adapter overhang sequences: forward (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′) and reverse (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′), following established Illumina 16S library preparation guidance. [1] Samples were multiplexed using the Nextera XT Index Kit (Illumina, #FC-131-1001) and sequenced by 500-cycle paired-end sequencing (2×250 bp) on an Illumina MiSeq (Illumina, #MS-102-2003). Bioinformatics processing and QC
Raw FASTQ files were inspected with FastQC. [2] Sequence processing was performed in QIIME 2. [3] Demultiplexed paired-end reads were imported using a Phred33 manifest and per-base quality profiles were reviewed to guide trimming/truncation. ASVs were inferred using the QIIME 2 DADA2 workflow. [4] Primer/adapter-proximal bases were removed using fixed 5′ trimming (17 bp forward; 21 bp reverse), reads were filtered using expected-error thresholds, and multiple forward/reverse truncation length combinations were evaluated to optimize read retention and paired-end merging for the ~460 bp V3–V4 amplicon. Final denoising parameters were selected based on denoising statistics (successful merging and proportion of non-chimeric reads across samples), and the resulting feature table and representative ASV sequences were used for downstream analyses.
Taxonomy was assigned by VSEARCH-based consensus classification [5] against SILVA (release 132) [6] at 97% identity. Downstream visualization and statistics were performed in R using phyloseq. [7] Diversity analyses, ordination, and statistics
Alpha diversity (Observed features/OTU richness, Shannon diversity, and Faith’s phylogenetic diversity) was calculated on rarefied data and compared between groups using nonparametric rank-based tests (Wilcoxon rank-sum for two-group comparisons; Kruskal–Wallis for ≥3 groups with post-hoc pairwise Wilcoxon tests). [8–10]
Group differences in community composition (beta-diversity) were assessed using Bray–Curtis dissimilarities [11] (computed after Hellinger transformation of abundance profiles) [12] and tested using PERMANOVA (adonis2 in vegan) with permutation-based significance. [13–14] Ordination was performed by principal coordinates analysis (PCoA) on the Bray–Curtis distance matrix to visualize sample-to-sample relationships. [15]
Differential abundance testing was performed using DESeq2 (negative binomial GLM). [16] Across analyses involving multiple hypothesis tests, p-values were adjusted using the Benjamini–Hochberg false discovery rate procedure. [17] Pathway inference
Predicted functional potential (gene families and pathways) was inferred from 16S ASVs using PICRUSt2. [18] References
Illumina. 16S Metagenomic Sequencing Library Preparation (Part #15044223 Rev. B) [Internet]. Illumina; 2013. (Includes the overhang adapter primer structure and V3–V4 primer sequences).
Andrews S. FastQC: a quality control tool for high throughput sequence data [Internet]. Babraham Bioinformatics; 2010 [cited 2025 Dec 18]. Available from: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi:10.1038/s41587-019-0209-9.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi:10.1038/nmeth.3869.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi:10.7717/peerj.2584.
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(D1):D590–D596. doi:10.1093/nar/gks1219.
McMurdie PJ, Holmes S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8(4):e61217. doi:10.1371/journal.pone.0061217.
Wilcoxon F. Individual comparisons by ranking methods. Biometrics Bulletin. 1945;1(6):80–83. doi:10.2307/3001968.
Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621. doi:10.1080/01621459.1952.10483441.
(If you want an additional general nonparametric/post-hoc reference beyond Wilcoxon/Kruskal–Wallis, tell me your preferred one and I’ll format it consistently.)
Bray JR, Curtis JT. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr. 1957;27:325–349. doi:10.2307/1942268.
Legendre P, Gallagher ED. Ecologically meaningful transformations for ordination of species data. Oecologia. 2001;129(2):271–280. doi:10.1007/s004420100716.
Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. doi:10.1111/j.1442-9993.2001.01070.pp.x.
Oksanen J, Simpson GL, Blanchet FG, Kindt R, Legendre P, Minchin PR, et al. vegan: Community Ecology Package [Internet]. R package. Available from: https://CRAN.R-project.org/package=vegan. doi:10.32614/CRAN.package.vegan.
Gower JC. Some distance properties of latent root and vector methods used in multivariate analysis. Biometrika. 1966;53(3–4):325–338. doi:10.1093/biomet/53.3-4.325.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi:10.1186/s13059-014-0550-8.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57(1):289–300. doi:10.1111/j.2517-6161.1995.tb02031.x.
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, et al. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38(6):685–688. doi:10.1038/s41587-020-0548-6. (slamo.biochem.dal.ca)Phyloseq_Group9_10_11_pre-FMT.Rmd
author: “”
date: ‘r format(Sys.time(), "%d %m %Y")‘
header-includes:
#install.packages(c("picante", "rmdformats"))
#mamba install -c conda-forge freetype libpng harfbuzz fribidi
#mamba install -c conda-forge r-systemfonts r-svglite r-kableExtra freetype fontconfig harfbuzz fribidi libpng
library(knitr)
library(rmdformats)
library(readxl)
library(dplyr)
library(kableExtra)
library(openxlsx)
library(DESeq2)
library(writexl)
options(max.print="75")
knitr::opts_chunk$set(fig.width=8,
fig.height=6,
eval=TRUE,
cache=TRUE,
echo=TRUE,
prompt=FALSE,
tidy=FALSE,
comment=NA,
message=FALSE,
warning=FALSE)
opts_knit$set(width=85)
#rmarkdown::render('Phyloseq_Group9_10_11_pre-FMT.Rmd',output_file='Phyloseq_Group9_10_11_pre-FMT.html')
# Phyloseq R library
#* Phyloseq web site : https://joey711.github.io/phyloseq/index.html
#* See in particular tutorials for
# - importing data: https://joey711.github.io/phyloseq/import-data.html
# - heat maps: https://joey711.github.io/phyloseq/plot_heatmap-examples.htmlImport raw data and assign sample key:
#extend qiime2_metadata_for_qza_to_phyloseq.tsv with Diet and Flora
#setwd("~/DATA/Data_Laura_16S_2/core_diversity_e4753")
#map_corrected <- read.csv("qiime2_metadata_for_qza_to_phyloseq.tsv", sep="\t", row.names=1)
#knitr::kable(map_corrected) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))install.packages("dplyr") # To manipulate dataframes
install.packages("readxl") # To read Excel files into R
install.packages("ggplot2") # for high quality graphics
install.packages("heatmaply")
source("https://bioconductor.org/biocLite.R")
biocLite("phyloseq")#mamba install -c conda-forge r-ggplot2 r-vegan r-data.table
#BiocManager::install("microbiome")
#install.packages("ggpubr")
#install.packages("heatmaply")
library("readxl") # necessary to import the data from Excel file
library("ggplot2") # graphics
library("picante")
library("microbiome") # data analysis and visualisation
library("phyloseq") # also the basis of data object. Data analysis and visualisation
library("ggpubr") # publication quality figures, based on ggplot2
library("dplyr") # data handling, filter and reformat data frames
library("RColorBrewer") # nice color options
library("heatmaply")
library(vegan)
library(gplots)
#install.packages("openxlsx")
library(openxlsx)Three tables are needed
library(tidyr)
# For QIIME1
#ps.ng.tax <- import_biom("./exported_table/feature-table.biom", "./exported-tree/tree.nwk")
# For QIIME2
#install.packages("remotes")
#remotes::install_github("jbisanz/qiime2R")
#"core_metrics_results/rarefied_table.qza", rarefying performed in the code, therefore import the raw table.
library(qiime2R)
ps_raw <- qza_to_phyloseq(
features = "table.qza", #cp ../Data_Karoline_16S_2025/dada2_tests2/test_7_f240_r240/table.qza .
tree = "rooted-tree.qza", #cp ../Data_Karoline_16S_2025/rooted-tree.qza .
metadata = "qiime2_metadata_for_qza_to_phyloseq.tsv" #cp ../Data_Karoline_16S_2025/qiime2_metadata_for_qza_to_phyloseq.tsv .
)
# or
#biom convert \
# -i ./exported_table/feature-table.biom \
# -o ./exported_table/feature-table-v1.biom \
# --to-json
#ps_raw <- import_biom("./exported_table/feature-table-v1.biom", treefilename="./exported-tree/tree.nwk")
sample <- read.csv("./qiime2_metadata_for_qza_to_phyloseq.tsv", sep="\t", row.names=1)
SAM = sample_data(sample, errorIfNULL = T)
#> setdiff(rownames(SAM), sample_names(ps_raw))
#[1] "sample-L9" should be removed since the low reads
ps_base <- merge_phyloseq(ps_raw, SAM)
print(ps_base)
taxonomy <- read.delim("taxonomy.tsv", sep="\t", header=TRUE) #cp ../Data_Karoline_16S_2025/exported-taxonomy/taxonomy.tsv .
#head(taxonomy)
# Separate taxonomy string into separate ranks
taxonomy_df <- taxonomy %>% separate(Taxon, into = c("Domain","Phylum","Class","Order","Family","Genus","Species"), sep = ";", fill = "right", extra = "drop")
# Use Feature.ID as rownames
rownames(taxonomy_df) <- taxonomy_df$Feature.ID
taxonomy_df <- taxonomy_df[, -c(1, ncol(taxonomy_df))] # Drop Feature.ID and Confidence
# Create tax_table
tax_table_final <- phyloseq::tax_table(as.matrix(taxonomy_df))
# Merge tax_table with existing phyloseq object
ps_base <- merge_phyloseq(ps_base, tax_table_final)
# Check
ps_base
#colnames(phyloseq::tax_table(ps_base)) <- c("Domain","Phylum","Class","Order","Family","Genus","Species")
saveRDS(ps_base, "./ps_base.rds")Visualize data
sample_names(ps_base)
rank_names(ps_base)
sample_variables(ps_base)
# Define sample names once
samples <- c(
#"sample-A1","sample-A2","sample-A5","sample-A6","sample-A7","sample-A8","sample-A9","sample-A10", #RESIZED: "sample-A3","sample-A4","sample-A11",
#"sample-B1","sample-B2","sample-B3","sample-B4","sample-B5","sample-B6","sample-B7", #RESIZED: "sample-B8","sample-B9","sample-B10","sample-B11","sample-B12","sample-B13","sample-B14","sample-B15","sample-B16",
#"sample-C1","sample-C2","sample-C3","sample-C4","sample-C5","sample-C6","sample-C7", #RESIZED: "sample-C8","sample-C9","sample-C10",
#"sample-E1","sample-E2","sample-E3","sample-E4","sample-E5","sample-E6","sample-E7","sample-E8","sample-E9","sample-E10", #RESIZED:
#"sample-F1","sample-F2","sample-F3","sample-F4","sample-F5",
"sample-G1","sample-G2","sample-G3","sample-G4","sample-G5","sample-G6",
"sample-H1","sample-H2","sample-H3","sample-H4","sample-H5","sample-H6",
"sample-I1","sample-I2","sample-I3","sample-I4","sample-I5","sample-I6",
"sample-J1","sample-J2","sample-J3","sample-J4","sample-J10","sample-J11", #RESIZED: "sample-J5","sample-J8","sample-J9", "sample-J6","sample-J7",
"sample-K1","sample-K2","sample-K3","sample-K4","sample-K5","sample-K6", #RESIZED: "sample-K7","sample-K8","sample-K9","sample-K10", "sample-K11","sample-K12","sample-K13","sample-K14","sample-K15",
"sample-L2","sample-L3","sample-L4","sample-L5","sample-L6" #RESIZED:"sample-L1","sample-L7","sample-L8","sample-L10","sample-L11","sample-L12","sample-L13","sample-L14","sample-L15",
#"sample-M1","sample-M2","sample-M3","sample-M4","sample-M5","sample-M6","sample-M7","sample-M8",
#"sample-N1","sample-N2","sample-N3","sample-N4","sample-N5","sample-N6","sample-N7","sample-N8","sample-N9","sample-N10",
#"sample-O1","sample-O2","sample-O3","sample-O4","sample-O5","sample-O6","sample-O7","sample-O8"
)
ps_pruned <- prune_samples(samples, ps_base)
sample_names(ps_pruned)
rank_names(ps_pruned)
sample_variables(ps_pruned)No samples were excluded as low-depth outliers (library sizes below the minimum depth threshold of 1,000 reads), and the remaining dataset (ps_filt) contains only samples meeting this depth cutoff with taxa retained only if they have nonzero total counts.
# ------------------------------------------------------------
# Filter low-depth samples (recommended for all analyses)
# ------------------------------------------------------------
min_depth <- 1000 # <-- adjust to your data / study design, keeps all!
ps_filt <- prune_samples(sample_sums(ps_pruned) >= min_depth, ps_pruned)
ps_filt <- prune_taxa(taxa_sums(ps_filt) > 0, ps_filt)
# Keep a depth summary for reporting / QC
depth_summary <- summary(sample_sums(ps_filt))
depth_summaryDifferential abundance (DESeq2) → ps_deseq: non-rarefied integer counts derived from ps_filt, with optional count-based taxon prefilter
(default: taxa total counts ≥ 10 across all samples)
From ps_filt (e.g. 5669 taxa and 239 samples), we branch into analysis-ready objects in two directions:
Alpha diversity: ps_rarefied ✅ (common)
Beta diversity:
ps_rarefied ✅ (often recommended)ps_rel or Hellinger ✅ (rarefaction optional)Normalize number of reads in each sample using median sequencing depth.
# RAREFACTION
set.seed(9242) # This will help in reproducing the filtering and nomalisation.
ps_rarefied <- rarefy_even_depth(ps_filt, sample.size = 6389)
#total <- 6389
# # NORMALIZE number of reads in each sample using median sequencing depth.
# total = median(sample_sums(ps.ng.tax))
# #> total
# #[1] 42369
# standf = function(x, t=total) round(t * (x / sum(x)))
# ps.ng.tax = transform_sample_counts(ps.ng.tax, standf)
# ps_rel <- microbiome::transform(ps.ng.tax, "compositional")
#
# saveRDS(ps.ng.tax, "./ps.ng.tax.rds")Taxonomic composition → ps_rel: relative abundance (compositional) computed after sample filtering (e.g. 5669 taxa and 239 samples)
Optional cleaner composition plots → ps_abund / ps_abund_rel: taxa filtered for plotting (e.g., keep taxa with mean relative abundance > 0.1%); (e.g. 95 taxa and 239 samples)
ps_abund = counts, ps_abund_rel = relative abundance (use for visualization, not DESeq2)
For the heatmaps, we focus on the most abundant OTUs by first converting counts to relative abundances within each sample. We then filter to retain only OTUs whose mean relative abundance across all samples exceeds 0.1% (0.001). We are left with 199 OTUs which makes the reading much more easy.
# 1) Convert to relative abundances
ps_rel <- transform_sample_counts(ps_filt, function(x) x / sum(x))
# 2) Get the logical vector of which OTUs to keep (based on relative abundance)
keep_vector <- phyloseq::filter_taxa(
ps_rel,
function(x) mean(x) > 0.001,
prune = FALSE
)
# 3) Use the TRUE/FALSE vector to subset absolute abundance data
ps_abund <- prune_taxa(names(keep_vector)[keep_vector], ps_filt)
# 4) Normalize the final subset to relative abundances per sample
ps_abund_rel <- transform_sample_counts(
ps_abund,
function(x) x / sum(x)
)library(stringr)
#for id in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206; do
#for id in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62; do
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Domain\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Domain\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Phylum\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Phylum\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Class\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Class\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Order\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Order\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Family\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Family\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Genus\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Genus\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Species\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Species\"], \"__\")[[1]][2]"
#done
phyloseq::tax_table(ps_abund_rel)[1,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[96,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[96,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[97,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[97,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[98,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[98,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[99,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[99,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[100,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[100,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[101,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[101,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[102,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[102,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[103,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[103,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[104,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[104,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[105,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[105,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[106,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[106,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[107,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[107,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[108,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[108,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[109,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[109,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[110,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[110,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[111,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[111,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[112,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[112,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[113,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[113,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[114,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[114,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[115,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[115,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[116,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[116,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[117,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[117,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[118,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[118,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[119,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[119,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[120,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[120,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[121,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[121,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[122,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[122,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[123,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[123,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[124,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[124,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[125,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[125,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[126,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[126,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[127,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[127,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[128,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[128,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[129,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[129,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[130,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[130,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[131,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[131,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[132,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[132,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[133,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[133,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[134,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[134,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[135,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[135,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[136,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[136,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[137,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[137,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[138,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[138,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[139,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[139,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[140,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[140,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[141,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[141,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[142,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[142,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[143,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[143,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[144,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[144,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[145,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[145,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[146,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[146,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[147,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[147,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[148,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[148,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[149,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[149,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[150,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[150,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[151,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[151,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[152,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[152,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[153,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[153,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[154,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[154,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[155,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[155,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[156,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[156,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[157,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[157,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[158,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[158,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[159,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[159,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[160,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[160,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[161,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[161,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[162,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[162,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[163,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[163,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[164,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[164,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[165,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[165,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[166,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[166,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[167,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[167,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[168,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[168,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[169,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[169,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[170,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[170,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[171,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[171,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[172,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[172,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[173,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[173,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[174,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[174,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[175,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[175,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[176,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[176,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[177,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[177,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[178,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[178,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[179,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[179,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[180,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[180,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[181,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[181,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[182,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[182,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[183,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[183,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[184,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[184,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[185,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[185,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[186,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[186,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[187,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[187,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[188,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[188,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[189,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[189,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[190,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[190,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[191,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[191,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[192,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[192,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[193,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[193,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[194,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[194,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[195,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[195,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[196,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[196,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[197,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[197,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[198,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[198,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[199,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[199,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[200,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[200,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[201,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[201,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[202,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[202,"Species"], "__")[[1]][2]
#aes(color="Phylum", fill="Phylum") --> aes()
#ggplot(data=data, aes(x=Sample, y=Abundance, fill=Phylum))
#options(max.print = 1e6)
my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, fill="Phylum") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=2)) #6 instead of theme.size\pagebreak Regroup together pre vs post stroke samples and normalize number of reads in each group using median sequencing depth.
ps_abund_rel_group <- merge_samples(ps_abund_rel, "G9_10_11_preFMT")
#PENDING: The effect weighted twice by sum(x), is the same to the effect weighted once directly from absolute abundance?!
ps_abund_rel_group_ = transform_sample_counts(ps_abund_rel_group, function(x) x / sum(x))
#plot_bar(ps_abund_relSampleType_, fill = "Phylum") + geom_bar(aes(color=Phylum, fill=Phylum), stat="identity", position="stack")
plot_bar(ps_abund_rel_group_, fill="Phylum") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, fill="Class") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=3))Regroup together pre vs post stroke samples and normalize number of reads in each group using median sequencing depth.
plot_bar(ps_abund_rel_group_, fill="Class") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)\pagebreak
my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, fill="Order") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=4))Regroup together pre vs post stroke and normalize number of reads in each group using median sequencing depth.
plot_bar(ps_abund_rel_group_, fill="Order") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)\pagebreak
my_colors <- c(
"#FF0000", "#000000", "#0000FF", "#C0C0C0", "#FFFFFF", "#FFFF00", "#00FFFF", "#FFA500", "#00FF00", "#808080", "#FF00FF", "#800080", "#FDD017", "#0000A0", "#3BB9FF", "#008000", "#800000", "#ADD8E6", "#F778A1", "#800517", "#736F6E", "#F52887", "#C11B17", "#5CB3FF", "#A52A2A", "#FF8040", "#2B60DE", "#736AFF", "#1589FF", "#98AFC7", "#8D38C9", "#307D7E", "#F6358A", "#151B54", "#6D7B8D", "#FDEEF4", "#FF0080", "#F88017", "#2554C7", "#FFF8C6", "#D4A017", "#306EFF", "#151B8D", "#9E7BFF", "#EAC117", "#E0FFFF", "#15317E", "#6C2DC7", "#FBB917", "#FCDFFF", "#15317E", "#254117", "#FAAFBE", "#357EC7"
)
plot_bar(ps_abund_rel, fill="Family") + geom_bar(aes(), stat="identity", position="stack") +
scale_fill_manual(values = my_colors) + theme(axis.text = element_text(size = 7, colour="black")) + theme(legend.position="bottom") + guides(fill=guide_legend(nrow=8))Regroup together pre vs post stroke samples and normalize number of reads in each group using median sequencing depth.
plot_bar(ps_abund_rel_group_, fill="Family") +
geom_bar(stat="identity", position="stack") +
scale_x_discrete(limits = c("pre-FMT", "Group9","Group10","Group11")) +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(angle = 30, hjust = 1, vjust = 1, size = 11, colour = "black"),
axis.text.y = element_text(angle = 0, size = 11, colour = "black"),
axis.title.x = element_text(size = 12, colour = "black"),
axis.title.y = element_text(size = 12, colour = "black")
)\pagebreak
\pagebreak
Plot Chao1 richness estimator, Observed OTUs, Shannon index, and Phylogenetic diversity. Regroup together samples from the same group.
# using rarefied data
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even42369.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering/rep_set.tre
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even4753.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering_stool/rep_set.tre
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even4753.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering_swab/rep_set.tre
library(dplyr)
library(reshape2)
library(ggpubr)
library(phyloseq)
library(kableExtra)
# ------------------------------------------------------------
# 0) Restrict analysis to the specified sample subset
# ------------------------------------------------------------
stopifnot(exists("samples"))
stopifnot(exists("ps_rarefied"))
ps_rarefied_sub <- prune_samples(samples, ps_rarefied)
hmp.meta <- meta(ps_rarefied_sub)
hmp.meta$sam_name <- rownames(hmp.meta)
# ------------------------------------------------------------
# 1) Read QIIME2 alpha-diversity outputs
# ------------------------------------------------------------
shannon <- read.table("exported_alpha/shannon/alpha-diversity.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
faith_pd <- read.table("exported_alpha/faith_pd/alpha-diversity.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
observed <- read.table("exported_alpha/observed_features/alpha-diversity.tsv",
header = TRUE, sep = "\t", stringsAsFactors = FALSE)
colnames(shannon) <- c("sam_name", "shannon")
colnames(faith_pd) <- c("sam_name", "PD_whole_tree")
colnames(observed) <- c("sam_name", "observed_otus")
div.df <- Reduce(function(x, y) merge(x, y, by = "sam_name", all = FALSE),
list(shannon, faith_pd, observed))
# ------------------------------------------------------------
# 2) Merge in metadata (from the pruned phyloseq object)
# ------------------------------------------------------------
div.df <- merge(div.df, hmp.meta, by = "sam_name", all.x = TRUE, all.y = FALSE)
# Keep ONLY samples from the predefined list (safety net)
div.df <- div.df %>% filter(sam_name %in% samples)
# ------------------------------------------------------------
# 3) Collapse groups: Group6/7/8 -> pre-FMT; keep only 4 groups
# ------------------------------------------------------------
div.df <- div.df %>%
mutate(
Group4 = case_when(
Group %in% c("Group6","Group7","Group8") ~ "pre-FMT",
Group %in% c("Group9","Group10","Group11") ~ as.character(Group),
TRUE ~ NA_character_
),
Group4 = factor(Group4, levels = c("pre-FMT","Group9","Group10","Group11"))
) %>%
filter(!is.na(Group4))
# ------------------------------------------------------------
# 4) Reformat table for reporting
# ------------------------------------------------------------
div.df2 <- div.df[, c("sam_name", "Group4", "shannon", "observed_otus", "PD_whole_tree")]
colnames(div.df2) <- c("Sample name", "Group", "Shannon", "OTU", "Phylogenetic Diversity")
write.csv(div.df2, file = "alpha_diversities_G9_10_11_preFMT.csv", row.names = FALSE)
#knitr::kable(div.df2) %>%
# kable_styling(bootstrap_options = c("striped","hover","condensed","responsive"))
# ------------------------------------------------------------
# 5) QC: print which samples were used
# ------------------------------------------------------------
requested_samples <- samples
present_in_ps <- sample_names(ps_rarefied)
missing_in_ps <- setdiff(requested_samples, present_in_ps)
cat("\n==== QC: Sample selection (ps_rarefied) ====\n")
cat("Requested samples:", length(requested_samples), "\n")
cat("Present in ps_rarefied:", length(present_in_ps), "\n")
cat("Missing from ps_rarefied:", length(missing_in_ps), "\n")
if (length(missing_in_ps) > 0) print(missing_in_ps)
used_samples <- div.df2$`Sample name`
missing_in_alpha <- setdiff(requested_samples, used_samples)
extra_in_alpha <- setdiff(used_samples, requested_samples)
cat("\n==== QC: Samples used in alpha-div df (div.df2) ====\n")
cat("Used samples:", length(used_samples), "\n")
cat("Requested but NOT used:", length(missing_in_alpha), "\n")
if (length(missing_in_alpha) > 0) print(missing_in_alpha)
cat("Used but NOT requested (should be 0):", length(extra_in_alpha), "\n")
if (length(extra_in_alpha) > 0) print(extra_in_alpha)
qc_table <- div.df %>%
select(sam_name, Group, Group4) %>%
distinct() %>%
arrange(Group4, Group, sam_name)
#cat("\n==== QC: Sample -> Group mapping used for plotting ====\n")
#print(qc_table)
#
#cat("\n==== QC: Counts per collapsed group (Group4) ====\n")
#print(div.df$Group4)
# ------------------------------------------------------------
# 6) Melt + plot
# ------------------------------------------------------------
div_df_melt <- reshape2::melt(
div.df2,
id.vars = c("Sample name","Group"),
variable.name = "variable",
value.name = "value"
)
# ensure final group order
div_df_melt$Group <- factor(div_df_melt$Group, levels = c("pre-FMT","Group9","Group10","Group11"))
#p <- ggboxplot(div_df_melt, x = "Group", y = "value",
# facet.by = "variable",
# scales = "free",
# width = 0.5,
# fill = "gray", legend = "right") +
# theme(
# axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1, size = 8),
# axis.text.y = element_text(size = 10),
# axis.title.x = element_text(size = 12),
# axis.title.y = element_text(size = 12)
# )
# all pairwise comparisons among the 4 groups
#lev <- levels(droplevels(div_df_melt$Group))
#L.pairs <- combn(lev, 2, simplify = FALSE)
#
#p2 <- p + stat_compare_means(
# method = "wilcox.test",
# comparisons = L.pairs,
# label = "p.signif",
# p.adjust.method = "BH",
# hide.ns = FALSE,
# symnum.args = list(
# cutpoints = c(0, 0.0001, 0.001, 0.01, 0.05, 1),
# symbols = c("****", "***", "**", "*", "ns")
# )
#)
#p2
# ------------------------------------------------------------
# 7) Save figure
# ------------------------------------------------------------
#ggsave("./figures/alpha_diversity_G9_10_11_preFMT.png", p2, device="png", height = 10, width = 15, dpi = 300)
#ggsave("./figures/alpha_diversity_G9_10_11_preFMT.svg", p2, device="svg", height = 10, width = 15)
# ------------------------------------------------------------
# 8) Save statistics
# ------------------------------------------------------------
library(dplyr)
library(purrr)
# same pair list as in the plot
lev <- levels(droplevels(div_df_melt$Group))
L.pairs <- combn(lev, 2, simplify = FALSE)
# Pairwise Wilcoxon per metric, with BH adjustment (matches p.adjust.method="BH")
stats_for_plot <- div_df_melt %>%
group_by(variable) %>%
group_modify(~{
dat <- .x
res <- purrr::map_dfr(L.pairs, function(pr) {
g1 <- pr[1]; g2 <- pr[2]
x1 <- dat$value[dat$Group == g1]
x2 <- dat$value[dat$Group == g2]
wt <- suppressWarnings(wilcox.test(x1, x2, exact = FALSE))
tibble(
group1 = g1,
group2 = g2,
n1 = length(x1),
n2 = length(x2),
p = unname(wt$p.value)
)
})
res %>%
mutate(
p.adj = p.adjust(p, method = "BH"),
p.signif = case_when(
p.adj <= 0.0001 ~ "****",
p.adj <= 0.001 ~ "***",
p.adj <= 0.01 ~ "**",
p.adj <= 0.05 ~ "*",
TRUE ~ "ns"
)
)
}) %>%
ungroup() %>%
rename(Metric = variable) %>%
arrange(Metric, group1, group2)
# Print the exact statistics used for annotation
print(stats_for_plot, n = Inf)
write.csv(stats_for_plot, "./figures/alpha_diversity_pairwise_stats_used_for_plot.csv", row.names = FALSE)
library(openxlsx)
# (optional) make sure the folder exists
dir.create("./figures", showWarnings = FALSE, recursive = TRUE)
out_xlsx <- "./figures/alpha_diversity_pairwise_stats_used_for_plot.xlsx"
write.xlsx(
x = list("pairwise_stats" = stats_for_plot),
file = out_xlsx,
overwrite = TRUE
)
cat("Saved Excel file to: ", out_xlsx, "\n", sep = "")# ---------------------------------------------------------------------------------
# ----------------------------- draw plots from input -----------------------------
library(dplyr)
library(ggpubr)
library(reshape2)
# Example: read from a CSV file you wrote earlier
# stats_for_plot <- read.csv("./figures/alpha_diversity_pairwise_stats_used_for_plot.csv", stringsAsFactors = FALSE)
# Or if you paste as text:
txt <- 'Metric,group1,group2,n1,n2,p,p.adj,p.signif
Shannon,Group10,Group11,6,5,1,1,ns
Shannon,Group9,Group10,6,6,0.297953061608168,0.595906123216336,ns
Shannon,Group9,Group11,6,5,0.522816653919089,0.631819188094748,ns
Shannon,pre-FMT,Group10,18,6,0.017949090591566,0.0538472717746981,ns
Shannon,pre-FMT,Group11,18,5,0.00651735384842052,0.0391041230905231,*
Shannon,pre-FMT,Group9,18,6,0.526515990078957,0.631819188094748,ns
OTU,Group10,Group11,6,5,0.143214612017615,0.214821918026422,ns
OTU,Group9,Group10,6,6,0.229766270461138,0.275719524553366,ns
OTU,Group9,Group11,6,5,0.522816653919089,0.522816653919089,ns
OTU,pre-FMT,Group10,18,6,0.0015274145728676,0.00916448743720559,**
OTU,pre-FMT,Group11,18,5,0.0675601586409843,0.135120317281969,ns
OTU,pre-FMT,Group9,18,6,0.00759484512604444,0.0227845353781333,*
Phylogenetic Diversity,Group10,Group11,6,5,1,1,ns
Phylogenetic Diversity,Group9,Group10,6,6,1,1,ns
Phylogenetic Diversity,Group9,Group11,6,5,0.927264473525232,1,ns
Phylogenetic Diversity,pre-FMT,Group10,18,6,0.000361550896811121,0.00108465269043336,**
Phylogenetic Diversity,pre-FMT,Group11,18,5,0.000910436385913111,0.00182087277182622,**
Phylogenetic Diversity,pre-FMT,Group9,18,6,0.000361550896811121,0.00108465269043336,**'
stats_for_plot <- read.csv(textConnection(txt), stringsAsFactors = FALSE)
# Match naming to your plot data: your facet variable is called "variable"
stats_for_plot <- stats_for_plot %>%
rename(variable = Metric) %>%
mutate(
group1 = factor(group1, levels = c("pre-FMT","Group9","Group10","Group11")),
group2 = factor(group2, levels = c("pre-FMT","Group9","Group10","Group11"))
)
# ------------------------------------------------------------
# Melt + plot (same as yours)
# ------------------------------------------------------------
div_df_melt <- reshape2::melt(
div.df2,
id.vars = c("Sample name","Group"),
variable.name = "variable",
value.name = "value"
)
div_df_melt$Group <- factor(div_df_melt$Group, levels = c("pre-FMT","Group9","Group10","Group11"))
p <- ggboxplot(div_df_melt, x = "Group", y = "value",
facet.by = "variable",
scales = "free",
width = 0.5,
fill = "gray", legend = "right") +
theme(
axis.text.x = element_text(angle = 45, hjust = 1, vjust = 1, size = 8),
axis.text.y = element_text(size = 10),
axis.title.x = element_text(size = 12),
axis.title.y = element_text(size = 12)
)
# ------------------------------------------------------------
# Add significance from your table (NO recomputation)
# ------------------------------------------------------------
# Compute y positions per facet so brackets don't overlap
ymax_by_metric <- div_df_melt %>%
group_by(variable) %>%
summarise(ymax = max(value, na.rm = TRUE), .groups = "drop")
stats_for_plot2 <- stats_for_plot %>%
left_join(ymax_by_metric, by = "variable") %>%
group_by(variable) %>%
arrange(p.adj) %>%
mutate(
# stack brackets within each facet
y.position = ymax * (1.05 + 0.08 * (row_number() - 1)),
# what to display (stars or "ns")
label = p.signif
) %>%
ungroup()
p2 <- p +
stat_pvalue_manual(
stats_for_plot2,
label = "label",
xmin = "group1",
xmax = "group2",
y.position = "y.position",
tip.length = 0.01,
hide.ns = FALSE
)
p2
# ------------------------------------------------------------
# Save
# ------------------------------------------------------------
ggsave("./figures/alpha_diversity_G9_10_11_preFMT_from_table.png",
p2, device="png", height = 6, width = 9, dpi = 300)
ggsave("./figures/alpha_diversity_G9_10_11_preFMT_from_table.svg",
p2, device="svg", height = 6, width = 9)
# label = sprintf("BH p=%.3g", p.adj)
Pairwise PERMANOVA tests were performed on Bray–Curtis distance matrices to compare bacterial community composition between all pairs of sample groups (metadata column Group). For each pairwise comparison, the distance matrix was subset to samples from the two groups only, and significance was assessed using vegan::adonis2 with 9,999 permutations. Resulting p-values were adjusted for multiple testing using both Benjamini–Hochberg (BH/FDR) and Bonferroni corrections.
Bray_pairwise_PERMANOVA <- read.csv("figures_MP_Group9_10_11_PreFMT/Bray_pairwise_PERMANOVA.csv", sep = ",")
knitr::kable(Bray_pairwise_PERMANOVA, caption = "Pairwise PERMANOVA results (distance-based community differences among Group levels).") %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))# --- Global beta-diversity (PERMANOVA) ---
cat("```text\n")
cat(
"[PERMANOVA result]\n",
"The object contained internal attribute: PCoA ADONIS\n",
"Permutation test for adonis under reduced model\n",
"Permutation: free\n",
"Number of permutations: 9999\n",
"\n",
"vegan::adonis2(formula = .formula, data = sampleda, permutations = permutations, method = distmethod)\n",
" Df SumOfSqs R2 F Pr(>F) \n",
"Model 3 5.3256 0.58559 14.602 1e-04 ***\n",
"Residual 31 3.7689 0.41441 \n",
"Total 34 9.0945 1.00000 \n",
"---\n",
"Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1\n",
sep = ""
)
cat("```\n")# --- Global beta-diversity (PERMANOVA) ---
cat("```text\n")
cat(
"[PERMANOVA result]\n",
"The object contained internal attribute: PCoA ADONIS\n",
"Permutation test for adonis under reduced model\n",
"Permutation: free\n",
"Number of permutations: 9999\n",
"\n",
"vegan::adonis2(formula = .formula, data = sampleda, permutations = permutations, method = distmethod)\n",
" Df SumOfSqs R2 F Pr(>F) \n",
"Model 2 0.82208 0.3324 3.4853 1e-04 ***\n",
"Residual 14 1.65109 0.6676 \n",
"Total 16 2.47317 1.0000 \n",
"---\n",
"Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1\n",
sep = ""
)
cat("```\n")knitr::include_graphics("figures_MP_Group9_10_11_PreFMT/PCoA3.png")knitr::include_graphics("figures_MP_Group9_10_11/PCoA3.png")Differential abundance analysis aims to find the differences in the abundance of each taxa between two groups of samples, assigning a significance value to each comparison.
# ------------------------------------------------------------
# DESeq2: non-rarefied integer counts + optional taxon prefilter
# ------------------------------------------------------------
ps_deseq <- ps_filt
ps_deseq_sel2 <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel2) <- otu_table(ps_deseq)[,c("sample-J1","sample-J2","sample-J3","sample-J4","sample-J10","sample-J11", "sample-K1","sample-K2","sample-K3","sample-K4","sample-K5","sample-K6")]
diagdds = phyloseq_to_deseq2(ps_deseq_sel2, ~Group)
diagdds$Group <- relevel(diagdds$Group, "Group9")
diagdds = DESeq(diagdds, test="Wald", fitType="parametric")
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel2)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group9_vs_Group10"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics("DEGs_Group9_vs_Group10.png")(Taxonomic dendrogram of microbial families filtered by relative abundance > 1%)
###############################################################################
# Taxonomic dendrogram of microbial families (abundance > 1%)
# Based on the Benakis et al. Figure 1C approach
# Author: [Your Name]
# Date: [Insert Date]
###############################################################################
# Load libraries
library(phyloseq)
library(dplyr)
library(tibble)
library(igraph)
library(ggraph)
# ------------------------------
# Step 1 — Agglomerate to Family level
# ------------------------------
# Assumes an existing phyloseq object: ps_filt
ps_fam <- tax_glom(ps_filt, taxrank = "Family", NArm = TRUE)
ps_fam <- prune_taxa(taxa_sums(ps_fam) > 0, ps_fam)
# Optional: restrict to selected groups if needed
# ps_fam <- subset_samples(ps_fam, Group %in% c("pre-FMT", "9", "10", "11"))
# ------------------------------
# Step 2 — Calculate relative abundance and filter families >1%
# ------------------------------
# Transform to relative abundance
ps_fam_relab <- transform_sample_counts(ps_fam, function(x) x / sum(x))
# Compute mean relative abundance across all samples
mean_abund <- taxa_sums(ps_fam_relab) / nsamples(ps_fam_relab)
# Keep only families with >1% mean relative abundance
keep_taxa <- names(mean_abund[mean_abund > 0.01])
ps_fam_top <- prune_taxa(keep_taxa, ps_fam_relab)
# ------------------------------
# Step 3 — Build taxonomic hierarchy (Phylum → Class → Order → Family)
# ------------------------------
tax <- as.data.frame(tax_table(ps_fam_top)) %>%
rownames_to_column("taxa_id") %>%
select(Phylum, Class, Order, Family) %>%
mutate(across(everything(), as.character)) %>%
distinct()
edges <- bind_rows(
tax %>% transmute(from = Phylum, to = Class),
tax %>% transmute(from = Class, to = Order),
tax %>% transmute(from = Order, to = Family)
) %>%
distinct() %>%
filter(!is.na(from), !is.na(to), from != "", to != "")
# ------------------------------
# Step 4 — Plot dendrogram-style taxonomy tree
# ------------------------------
g <- graph_from_data_frame(edges, directed = TRUE)
p <- ggraph(g, layout = "dendrogram") +
geom_edge_diagonal(color = "grey60") +
geom_node_point(size = 2, color = "steelblue") +
geom_node_text(aes(label = name), hjust = -0.1, size = 3) +
theme_void() +
coord_flip() +
ggtitle("Taxonomic dendrogram of microbial families (>1% abundance)")
print(p)
# ------------------------------
# Optional: collapse remaining rare families as "Other"
# ------------------------------
# This can be done before filtering or directly in the figure annotation
###############################################################################A taxonomic dendrogram illustrating the microbial composition at the family level was generated in R (version 4.3.0) using the phyloseq and ggraph packages. The analysis followed an approach similar to that presented by Benakis et al. (2020, Cell Host \& Microbe, Figure 1C).
First, the filtered amplicon sequence variant (ASV) table was agglomerated to the Family rank using the tax_glom() function from phyloseq. The dataset was then transformed to relative abundances for each sample, and families with a mean relative abundance greater than 1% across all samples were retained for visualization. Families below this threshold were excluded and considered part of the “Other” category for interpretability.
A hierarchical taxonomy table (Phylum → Class → Order → Family) was extracted, and edges representing parent–child relationships were assembled using dplyr. The resulting taxonomic relationships were converted into a directed graph structure using igraph, and visualized as a dendrogram with ggraph (layout = "dendrogram").
This approach provides a qualitative overview of microbial family-level diversity while reducing visual noise from rare taxa, consistent with the display strategy used in the original publication.
Would you like me to adapt the Methods version to a more formal journal submission style (e.g., third person passive voice and APA-compliant citation format)?
Concept overview
Yes—this is doable in R, and you can restrict the dendrogram to only significant microbial families. Based on the structure in the Benakis et al. paper:
If your PI asks for “only significantly expressed families,” you first need to perform a differential abundance test at the Family level (e.g., using ANCOM-BC2 or DESeq2) before plotting only those families or highlighting them on the tree.
library(phyloseq)
ps_fam <- tax_glom(ps_filt, taxrank = "Family", NArm = TRUE)
ps_fam <- prune_taxa(taxa_sums(ps_fam) > 0, ps_fam)
# Restrict to requested sample groups
ps_fam_sub <- subset_samples(ps_fam, Group %in% c("9", "10", "11", "pre-FMT"))
sample_data(ps_fam_sub)$Group <- factor(sample_data(ps_fam_sub)$Group,
levels = c("pre-FMT", "9", "10", "11"))Option A: ANCOM-BC2 (recommended for microbiome data)
library(ANCOMBC)
an2 <- ancombc2(data = ps_fam_sub,
formula = "Group",
p_adj_method = "BH",
prv_cut = 0, lib_cut = 0,
group = "Group",
global = FALSE)
res <- an2$res
# Inspect results: names(an2$res), matrices such as res$diff_abn and res$q_valOption B: DESeq2 (alternative)
library(DESeq2)
dds <- phyloseq_to_deseq2(ps_fam_sub, ~ Group)
dds <- DESeq(dds)
# Example contrast: Group 9 vs 10
r_9_10 <- results(dds, contrast = c("Group", "9", "10"))
sig_9_10 <- rownames(r_9_10)[which(r_9_10$padj < 0.05)]
sig_9_10 <- sig_9_10[!is.na(sig_9_10)]Decide what “significant” means for your plot:
library(dplyr)
library(tibble)
library(igraph)
library(ggraph)
# Choose your significant taxa list (example from DESeq2)
sig_taxa <- sig_9_10
# Get taxonomy for significant families
tax <- as.data.frame(tax_table(ps_fam_sub)) %>%
rownames_to_column("taxa_id") %>%
filter(taxa_id %in% sig_taxa) %>%
select(Phylum, Class, Order, Family) %>%
mutate(across(everything(), as.character)) %>%
distinct()
# Build hierarchical edges
edges <- bind_rows(
tax %>% transmute(from = Phylum, to = Class),
tax %>% transmute(from = Class, to = Order),
tax %>% transmute(from = Order, to = Family)
) %>%
distinct() %>%
filter(!is.na(from), !is.na(to), from != "", to != "")
# Build graph
g <- graph_from_data_frame(edges, directed = TRUE)
# Plot dendrogram
p <- ggraph(g, layout = "dendrogram") +
geom_edge_diagonal() +
geom_node_point(size = 2) +
geom_node_text(aes(label = name), hjust = -0.1, size = 3) +
theme_void() +
coord_flip()
pTo color families by direction (e.g., enriched in Group 9 vs 10), merge the log2FoldChange results from DESeq2 with the node names and use scale_color_gradient2() or similar in ggraph to map significance.
Yes—this can be done in R. The template figure in the Benakis paper shows a taxonomy-based dendrogram (Phylum → Class → Order → Family). In that study, families were filtered by abundance (only > 1% frequency) rather than statistical significance.
For our dataset, we can (i) construct the full family-level taxonomy tree and (ii) optionally restrict or highlight only those families that are significantly different between groups based on differential abundance testing (using DESeq2 or ANCOM-BC2 with Benjamini–Hochberg-adjusted p-values).
If you specify which comparison defines “significant” (only 9 vs 10 or across all four groups), the exact filtering and visualization code can be tailored to more closely match Figure 1C, including the “Other” bin and legend style.
## =========================================================
## MicrobiotaProcess_UPDATED.R
## Uses your objects:
## - ps_filt : for alpha + beta diversity (NOT taxa-filtered)
## - ps_abund_rel : for cleaner composition plots (taxa filtered for plotting)
##
## Output:
## - Rarefaction curves + alpha diversity plots
## - Bray (Hellinger) distance + PCoA + PERMANOVA
## - Composition plots (Class) + heatmaps (from ps_abund_rel)
## =========================================================
## -----------------------------
## 0) Define groups (sample IDs) ---- EXACTLY as provided
Group1<-c("U24080201","U25020701","O23092004","U24101801","U25022101","O23102703","A24062801","O23112205","U23071901","A23112002","U24111801","O23110101","U24121801","O23120101","O24011202","O23090803","A23060602","A24030402","U25011701","O24011901","U23090801","O24011201","O24011003","O23092202","O23082301","O23091403","O23112901","O23092201","O24013103","O24021403","O24010402","O23092005","O23092203","O24010302","O23090701","O23091501","O23092701","O24022202","O23092802","O23090601","O23100401","O24022102","O23081801","O23092006","O23100503","O23090602","O24013104","O24020803","O24010301","O24010404","O23090802","O23092801","O24022801","O23100706","O23102602","O24021601","O24012401","O24021603","O24022901","O24021501","O23110902","O23102601","O23102704","O23100803","O23102701","O24021401","O24022101","O24030603","O23110901","O23110903","O23110301","O24022301","O23102502","O23111501","O23111602","O24020705","O24021502","O24022201","O23110202","O23090801")
Group2<-c("U23071701","U23052401","U23052201","U24070401","O24011801","O23092003","A24071901","A24072901","O24011102","O23121501","O23092104","O23092001","O23121301","O24020701","O23112201","O23100701","O23100801","O24020903","O24020901","O24020703","O23112204")
Group3<-c("O23100802","O24011205","O23092002","O24011207","O23092103","O23102501","O24011005","A24030401","O24011004","A23051102","U25011702","O24011204","O23121502","O23120702","O24011206","O24021404","O23092101","O24010403","O23112303","O23083001","O23082302","O24010401","O24022302","O24010501","O23112902","O23082303","O23083102","O24013101","O23100402","O24020801","O23120701","O23121304","O24021602","O24011802","O23121306","O23120103","O24020905","O24012403","O24013102","O24021503","O24020904","O23102504","O24013105","O24030601","O23100705","O24030604","O23111601","O24020103","O24030602","O23110302","O23102603","O24031304","O24021402","O24020101","O24012501","O24020804","O23100804","O23102503","O24022902","O24020704","O23110904","O24020102","O24012402","O23102702","O23102604","O23110204","O23110203","O23083101","O23092702")
Group4<-c("O23112304","A23051103","A24071701","A23080101","A24031201","A24080201","O24011105","O23091305","O23121302","O23092803")
Group5<-c("O23091303","O23112301","O24011203","A23112001","O24011001","O24011002","O23091302","O24020902","O23092102","O23091401","O23121503","O23091402","O24020702","O23091301","O23112206","O24011103","O23121305","O24011101","O23121303","O24011104","O23120104","O23100704","O23112302","O23112203","O23100703","O24020805","O24020802","O23112202","O24031302","O23111502","O23100702","O24031301","O24031305","O23082402")
NTC<-c("NTC_2","NTC_3","NTC_16","NTC_4","NTC_6","NTC_7","NTC_8","NTC_9","NTC_11","NTC_12","NTC_13","NTC_14","NTC_15","NTC_10")
PC<-c("PC_1","PC_2","PC_8","PC_4","PC_3","PC_5","PC_6","PC_7","UR009768","UR009909","PC01")
## Ensure unique IDs within each vector
Group1 <- unique(Group1); Group2 <- unique(Group2); Group3 <- unique(Group3)
Group4 <- unique(Group4); Group5 <- unique(Group5); NTC <- unique(NTC); PC <- unique(PC)
## The group order you want everywhere (plots + stats)
group_levels <- c("Group1","Group2","Group3","Group4","Group5","NTC","PC")
## -----------------------------
## 0.1) Sample subset (union of all groups)
keep_samples <- unique(c(Group1, Group2, Group3, Group4, Group5, NTC, PC))
## -----------------------------
## 0.2) Helper: assign Group as an ordered factor (membership-based)
add_group <- function(mpse_obj) {
sn <- rownames(colData(mpse_obj))
grp <- rep(NA_character_, length(sn))
grp[sn %in% Group1] <- "Group1"
grp[sn %in% Group2] <- "Group2"
grp[sn %in% Group3] <- "Group3"
grp[sn %in% Group4] <- "Group4"
grp[sn %in% Group5] <- "Group5"
grp[sn %in% NTC] <- "NTC"
grp[sn %in% PC] <- "PC"
colData(mpse_obj)$Group <- factor(grp, levels = group_levels)
# warn if any samples weren't assigned (helps catch typos / missing IDs)
if (any(is.na(colData(mpse_obj)$Group))) {
warning(
"Unassigned samples (not found in any group list): ",
paste(sn[is.na(colData(mpse_obj)$Group)], collapse = ", ")
)
}
mpse_obj
}
## -----------------------------
## 0.3) Colors (edit to taste)
group_colors <- c(
"Group1" = "#1f77b4",
"Group2" = "#ff7f0e",
"Group3" = "#2ca02c",
"Group4" = "#d62728",
"Group5" = "#9467bd",
"NTC" = "#7f7f7f",
"PC" = "#8c564b"
)
## =========================================================
## 1) Diversity analysis object (alpha + beta)
## IMPORTANT: start from ps_filt (all taxa retained)
## =========================================================
ps_div <- prune_samples(keep_samples, ps_filt)
ps_div <- prune_taxa(taxa_sums(ps_div) > 0, ps_div)
mpse_div <- as.MPSE(ps_div)
mpse_div <- add_group(mpse_div)
cat("\n[mpse_div] Group counts:\n")
print(table(colData(mpse_div)$Group, useNA = "ifany"))
## =========================================================
## 2) Alpha diversity (rarefaction-based)
## =========================================================
set.seed(9242)
mpse_div %<>% mp_rrarefy() # creates RareAbundance
mpse_div %<>% mp_cal_rarecurve(.abundance = RareAbundance, chunks = 400)
# Rarefaction curves: sample + grouped
p_rare_1 <- mpse_div %>% mp_plot_rarecurve(.rare = RareAbundanceRarecurve, .alpha = Observe)
p_rare_2 <- mpse_div %>%
mp_plot_rarecurve(.rare = RareAbundanceRarecurve, .alpha = Observe, .group = Group) +
scale_color_manual(values = group_colors, guide = "none") +
scale_fill_manual(values = group_colors, guide = "none")
p_rare_3 <- mpse_div %>%
mp_plot_rarecurve(.rare = RareAbundanceRarecurve, .alpha = "Observe",
.group = Group, plot.group = TRUE) +
scale_color_manual(values = group_colors, guide = "none") +
scale_fill_manual(values = group_colors, guide = "none")
png("figures/rarefaction_of_samples_or_groups.png", width = 1080, height = 600)
print(p_rare_1 + p_rare_2 + p_rare_3)
dev.off()
# Alpha indices from RareAbundance
mpse_div %<>% mp_cal_alpha(.abundance = RareAbundance)
f_alpha_1 <- mpse_div %>%
mp_plot_alpha(.group = Group, .alpha = c(Observe, Chao1, ACE, Shannon, Simpson, Pielou)) +
scale_color_manual(values = group_colors, guide = "none") +
scale_fill_manual(values = group_colors, guide = "none")
f_alpha_2 <- mpse_div %>%
mp_plot_alpha(.alpha = c(Observe, Chao1, ACE, Shannon, Simpson, Pielou))
png("figures/alpha_diversity_comparison.png", width = 1400, height = 600)
print(f_alpha_1 / f_alpha_2)
dev.off()
## =========================================================
## 3) Beta diversity (Bray–Curtis on Hellinger)
## IMPORTANT: use non-rarefied Abundance (not taxa-filtered)
## =========================================================
mpse_div %<>% mp_decostand(.abundance = Abundance) # creates 'hellinger'
mpse_div %<>% mp_cal_dist(.abundance = hellinger, distmethod = "bray")
# Distance between samples
p_dist_1 <- mpse_div %>% mp_plot_dist(.distmethod = bray)
png("figures/distance_between_samples.png", width = 1000, height = 1000)
print(p_dist_1)
dev.off()
# Distance with group info
p_dist_2 <- mpse_div %>% mp_plot_dist(.distmethod = bray, .group = Group) +
scale_fill_manual(values = group_colors) +
scale_color_gradient()
png("figures/distance_between_samples_with_group_info.png", width = 1000, height = 1000)
print(p_dist_2)
dev.off()
# Compare distances between groups (Bray)
p_dist_3 <- mpse_div %>%
mp_plot_dist(.distmethod = bray, .group = Group, group.test = TRUE, textsize = 6) +
theme(
axis.title.x = element_text(size = 14),
axis.title.y = element_text(size = 14),
axis.text.x = element_text(size = 14),
axis.text.y = element_text(size = 14)
)
png("figures/Comparison_of_Bray_Distances.png", width = 1000, height = 1000)
print(p_dist_3)
dev.off()
## PCoA + PERMANOVA (adonis2)
mpse_div %<>% mp_cal_pcoa(.abundance = hellinger, distmethod = "bray")
mpse_div %<>% mp_adonis(
.abundance = hellinger,
.formula = ~ Group,
distmethod = "bray",
permutations = 9999,
action = "add"
)
cat("\n[PERMANOVA result]\n")
print(mpse_div %>% mp_extract_internal_attr(name = "adonis"))
## PCoA plot
p_pcoa_1 <- mpse_div %>%
mp_plot_ord(
.ord = pcoa,
.group = Group,
.color = Group,
.size = 4,
.alpha = 1,
ellipse = TRUE,
show.legend = TRUE
) +
scale_fill_manual(values = group_colors) +
scale_color_manual(values = group_colors) +
theme(
axis.text = element_text(size = 16),
axis.title = element_text(size = 18),
legend.text = element_text(size = 14),
legend.title= element_text(size = 16)
)
png("figures/PCoA.png", width = 1200, height = 1000)
print(p_pcoa_1)
dev.off()
pdf("figures/PCoA.pdf", width = 10, height = 8)
print(p_pcoa_1)
dev.off()
## Optional: label points
colData(mpse_div)$ShortLabel <- gsub("sample-", "", rownames(colData(mpse_div)))
p_pcoa_2 <- p_pcoa_1 +
geom_text_repel(aes(label = ShortLabel), size = 4, max.overlaps = 100)
png("figures/PCoA_labeled.png", width = 1200, height = 1000)
print(p_pcoa_2)
dev.off()
#[PERMANOVA result]
#The object contained internal attribute: PCoA ADONIS
#Permutation test for adonis under reduced model
#Permutation: free
#Number of permutations: 9999
#
#vegan::adonis2(formula = .formula, data = sampleda, permutations = permutations, method = distmethod)
# Df SumOfSqs R2 F Pr(>F)
#Model 6 11.446 0.16058 7.3971 1e-04 ***
#Residual 232 59.829 0.83942
#Total 238 71.274 1.00000
#---
#Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1
## =========================================================
## 4) Composition plots object (clean taxa set for plotting)
## IMPORTANT: start from ps_abund_rel (plotting-filtered taxa)
## =========================================================
ps_plot <- prune_samples(keep_samples, ps_abund_rel)
ps_plot <- prune_taxa(taxa_sums(ps_plot) > 0, ps_plot)
mpse_plot <- as.MPSE(ps_plot)
mpse_plot <- add_group(mpse_plot)
## Summaries for plotting (Class)
mpse_plot %<>%
mp_cal_abundance(.abundance = Abundance) %>% # per sample
mp_cal_abundance(.abundance = Abundance, .group = Group) # per group
## Class abundance barplots (top 20)
p_class_rel <- mpse_plot %>%
mp_plot_abundance(
.abundance = Abundance,
taxa.class = Class,
topn = 20,
relative = TRUE
)
p_class_abs <- mpse_plot %>%
mp_plot_abundance(
.abundance = Abundance,
taxa.class = Class,
topn = 20,
relative = FALSE
)
png("figures/relative_abundance_and_abundance_samples.png", width = 1200, height = 600)
print(p_class_rel / p_class_abs)
dev.off()
## Heatmaps (grouped)
h_rel <- mpse_plot %>%
mp_plot_abundance(
.abundance = Abundance,
.group = Group,
taxa.class = Class,
relative = TRUE,
topn = 20,
geom = "heatmap",
features.dist = "euclidean",
features.hclust = "average",
sample.dist = "bray",
sample.hclust = "average"
)
h_abs <- mpse_plot %>%
mp_plot_abundance(
.abundance = Abundance,
.group = Group,
taxa.class = Class,
relative = FALSE,
topn = 20,
geom = "heatmap",
features.dist = "euclidean",
features.hclust = "average",
sample.dist = "bray",
sample.hclust = "average"
)
png("figures/relative_abundance_and_abundance_heatmap.png", width = 1200, height = 600)
print(aplot::plot_list(gglist = list(h_rel, h_abs), tag_levels = "A"))
dev.off()
## Group-level barplots
p_group_rel <- mpse_plot %>%
mp_plot_abundance(
.abundance = Abundance,
.group = Group,
taxa.class = Class,
topn = 20,
plot.group = TRUE,
relative = TRUE
) +
scale_fill_manual(values = group_colors)
p_group_abs <- mpse_plot %>%
mp_plot_abundance(
.abundance = Abundance,
.group = Group,
taxa.class = Class,
topn = 20,
plot.group = TRUE,
relative = FALSE
) +
scale_fill_manual(values = group_colors)
png("figures/relative_abundance_and_abundance_groups.png", width = 1000, height = 1000)
print(p_group_rel / p_group_abs)
dev.off()
cat("\nDONE. Outputs written to ./figures/\n")
## =========================================================
## CONTINUE: Export Bray distances + pairwise PERMANOVA
## (Use mpse_div from the updated script above)
## =========================================================
suppressPackageStartupMessages({
library(dplyr)
library(tidyr)
library(openxlsx)
library(vegan)
})
## -----------------------------
## Helper: get assay matrix with rows = samples, cols = features
get_sample_by_feature <- function(mpse_obj, assay_name) {
mat <- assay(mpse_obj, assay_name)
# sample IDs in MPSE
samp_ids <- rownames(colData(mpse_obj))
# If samples are columns, transpose
if (!is.null(colnames(mat)) && all(samp_ids %in% colnames(mat))) {
mat <- t(mat)
}
# Now enforce row order to match colData
mat <- mat[samp_ids, , drop = FALSE]
mat
}
## -----------------------------
## 1) Recompute Bray–Curtis distance (robust extraction)
hell_mat_sf <- get_sample_by_feature(mpse_div, "hellinger") # rows=samples, cols=features
bray_dist <- vegdist(hell_mat_sf, method = "bray")
## sanity checks
stopifnot(!any(is.na(as.matrix(bray_dist))))
stopifnot(!any(as.matrix(bray_dist) < 0, na.rm = TRUE))
## -----------------------------
## 2) Export all pairwise Bray distances to Excel
bray_mat <- as.matrix(bray_dist)
samples <- rownames(bray_mat)
bray_df <- as.data.frame(as.table(bray_mat)) %>%
rename(Sample1 = Var1, Sample2 = Var2, BrayDistance = Freq) %>%
filter(Sample1 < Sample2) %>%
arrange(Sample1, Sample2)
write.xlsx(bray_df, file = "figures/Bray_Curtis_Distances.xlsx")
## -----------------------------
## 3) Pairwise PERMANOVA (post-hoc) using vegan::adonis2
meta2 <- as.data.frame(colData(mpse_div))
meta2 <- meta2[rownames(hell_mat_sf), , drop = FALSE]
meta2$Group <- droplevels(meta2$Group)
groups <- levels(meta2$Group)
res_list <- list()
k <- 1
for (i in 1:(length(groups) - 1)) {
for (j in (i + 1):length(groups)) {
g1 <- groups[i]
g2 <- groups[j]
idx <- meta2$Group %in% c(g1, g2)
sub_meta <- meta2[idx, , drop = FALSE]
sub_dist <- as.dist(as.matrix(bray_dist)[idx, idx])
ad <- adonis2(sub_dist ~ Group, data = sub_meta, permutations = 9999)
res_list[[k]] <- data.frame(
group1 = g1,
group2 = g2,
F = ad$F[1],
R2 = ad$R2[1],
p = ad$`Pr(>F)`[1]
)
k <- k + 1
}
}
pair_res <- do.call(rbind, res_list)
pair_res$p_adj_BH <- p.adjust(pair_res$p, method = "BH")
pair_res$p_adj_Bonferroni <- p.adjust(pair_res$p, method = "bonferroni")
write.csv(pair_res, "figures/Bray_pairwise_PERMANOVA.csv", row.names = FALSE)
cat("\nPairwise PERMANOVA written to: figures/Bray_pairwise_PERMANOVA.csv\n")
cat("Bray distance table written to: figures/Bray_Curtis_Distances.xlsx\n")
## =========================================================
## OPTIONAL: PCoA plot where point size = Shannon and alpha = Observe
## (requires mpse_div already has Shannon/Observe from mp_cal_alpha)
## =========================================================
p_pcoa_sizealpha <- mpse_div %>%
mp_plot_ord(
.ord = pcoa,
.group = Group,
.color = Group,
.size = Shannon,
.alpha = Observe,
ellipse = TRUE,
show.legend = TRUE
) +
scale_fill_manual(values = group_colors) +
scale_color_manual(values = group_colors) +
scale_size_continuous(range = c(2, 6)) +
theme(
axis.text = element_text(size = 16),
axis.title = element_text(size = 18),
legend.text = element_text(size = 14),
legend.title= element_text(size = 16)
)
png("figures/PCoA_sizealpha.png", width = 1200, height = 1000)
print(p_pcoa_sizealpha)
dev.off()
pdf("figures/PCoA_sizealpha.pdf", width = 10, height = 8)
print(p_pcoa_sizealpha)
dev.off()
## =========================================================
## Ensure all three ordination outputs exist:
## - PCoA : basic (color/group)
## - PCoA2 : size = Shannon, alpha = Observe
## - PCoA3 : same as PCoA2 + sample labels
##
## Assumes you already have:
## - mpse_div with: pcoa, Group, Shannon, Observe
## - group_colors defined
## =========================================================
p1 <- mpse_div %>%
mp_plot_ord(
.ord = pcoa,
.group = Group,
.color = Group,
.size = 4,
.alpha = 1,
ellipse = TRUE,
show.legend = FALSE
) +
scale_fill_manual(
values = group_colors,
guide = guide_legend(
keywidth = 1.6,
keyheight = 1.6,
label.theme = element_text(size = 16)
)
) +
scale_color_manual(
values = group_colors,
guide = guide_legend(
keywidth = 1.6,
keyheight = 1.6,
label.theme = element_text(size = 16)
)
) +
theme(
axis.text = element_text(size = 20),
axis.title = element_text(size = 22),
legend.text = element_text(size = 20),
legend.title= element_text(size = 22),
plot.title = element_text(size = 24, face = "bold"),
plot.subtitle = element_text(size = 20)
)
png("PCoA.png", width = 1200, height = 1000)
p1
dev.off()
pdf("PCoA.pdf")
p1
dev.off()
p2 <- mpse_div %>%
mp_plot_ord(
.ord = pcoa,
.group = Group,
.color = Group,
.size = Shannon,
.alpha = Observe,
ellipse = TRUE,
show.legend = FALSE
) +
scale_fill_manual(
values = group_colors,
guide = guide_legend(
keywidth = 0.6,
keyheight = 0.6,
label.theme = element_text(size = 16)
)
) +
scale_color_manual(
values = group_colors,
guide = guide_legend(
keywidth = 0.6,
keyheight = 0.6,
label.theme = element_text(size = 16)
)
) +
scale_size_continuous(
range = c(2, 6),
guide = guide_legend(
keywidth = 0.6,
keyheight = 0.6,
label.theme = element_text(size = 16)
)
) +
theme(
axis.text = element_text(size = 20),
axis.title = element_text(size = 22),
legend.text = element_text(size = 20),
legend.title= element_text(size = 22),
plot.title = element_text(size = 24, face = "bold"),
plot.subtitle = element_text(size = 20)
)
png("PCoA2.png", width = 1200, height = 1000)
p2
dev.off()
pdf("PCoA2.pdf")
p2
dev.off()
library(ggrepel)
colData(mpse_div)$ShortLabel <- gsub("sample-", "", mpse_div@colData@rownames)
p3 <- mpse_div %>%
mp_plot_ord(
.ord = pcoa,
.group = Group,
.color = Group,
.size = Shannon,
.alpha = Observe,
ellipse = TRUE,
show.legend = FALSE
) +
geom_text_repel(aes(label = ShortLabel), size = 5, max.overlaps = 100) +
scale_fill_manual(
values = group_colors,
guide = guide_legend(
keywidth = 0.6,
keyheight = 0.6,
label.theme = element_text(size = 16)
)
) +
scale_color_manual(
values = group_colors,
guide = guide_legend(
keywidth = 0.6,
keyheight = 0.6,
label.theme = element_text(size = 16)
)
) +
scale_size_continuous(
range = c(2, 6),
guide = guide_legend(
keywidth = 0.6,
keyheight = 0.6,
label.theme = element_text(size = 16)
)
) +
theme(
axis.text = element_text(size = 20),
axis.title = element_text(size = 22),
legend.text = element_text(size = 20),
legend.title= element_text(size = 22),
plot.title = element_text(size = 24, face = "bold"),
plot.subtitle = element_text(size = 20)
)
png("PCoA3.png", width = 1200, height = 1000)
p3
dev.off()
svg("PCoA3.svg", width = 12, height = 10)
p3
dev.off()
pdf("PCoA3.pdf")
p3
dev.off()author: ""
date: '`r format(Sys.time(), "%d %m %Y")`'
header-includes:
- \usepackage{color, fancyvrb}
output:
rmdformats::readthedown:
highlight: kate
number_sections : yes
pdf_document:
toc: yes
toc_depth: 2
number_sections : yes
---
```{r load-packages, include=FALSE}
#install.packages(c("picante", "rmdformats"))
#mamba install -c conda-forge freetype libpng harfbuzz fribidi
#mamba install -c conda-forge r-systemfonts r-svglite r-kableExtra freetype fontconfig harfbuzz fribidi libpng
library(knitr)
library(rmdformats)
library(readxl)
library(dplyr)
library(kableExtra)
library(openxlsx)
library(DESeq2)
library(writexl)
options(max.print="75")
knitr::opts_chunk$set(fig.width=8,
fig.height=6,
eval=TRUE,
cache=TRUE,
echo=TRUE,
prompt=FALSE,
tidy=FALSE,
comment=NA,
message=FALSE,
warning=FALSE)
opts_knit$set(width=85)
# Phyloseq R library
#* Phyloseq web site : https://joey711.github.io/phyloseq/index.html
#* See in particular tutorials for
# - importing data: https://joey711.github.io/phyloseq/import-data.html
# - heat maps: https://joey711.github.io/phyloseq/plot_heatmap-examples.html
#rmarkdown::render('Phyloseq.Rmd',output_file='Phyloseq.html')
#options(max.print = 1e6)
```
# Data
Import raw data and assign sample key:
```{r, echo=FALSE, warning=FALSE}
#extend qiime2_metadata_for_qza_to_phyloseq.tsv with Diet and Flora
#setwd("~/DATA/Data_Laura_16S_2/core_diversity_e4753")
#map_corrected <- read.csv("qiime2_metadata_for_qza_to_phyloseq.tsv", sep="\t", row.names=1)
#knitr::kable(map_corrected) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
```
# Prerequisites to be installed
* R : https://pbil.univ-lyon1.fr/CRAN/
* R studio : https://www.rstudio.com/products/rstudio/download/#download
```R
install.packages("dplyr") # To manipulate dataframes
install.packages("readxl") # To read Excel files into R
install.packages("ggplot2") # for high quality graphics
install.packages("heatmaply")
source("https://bioconductor.org/biocLite.R")
biocLite("phyloseq")
```
```{r libraries, echo=TRUE, message=FALSE}
#mamba install -c conda-forge r-ggplot2 r-vegan r-data.table
#BiocManager::install("microbiome")
#install.packages("ggpubr")
#install.packages("heatmaply")
library("readxl") # necessary to import the data from Excel file
library("ggplot2") # graphics
library("picante")
library("microbiome") # data analysis and visualisation
library("phyloseq") # also the basis of data object. Data analysis and visualisation
library("ggpubr") # publication quality figures, based on ggplot2
library("dplyr") # data handling, filter and reformat data frames
library("RColorBrewer") # nice color options
library("heatmaply")
library(vegan)
library(gplots)
#install.packages("openxlsx")
library(openxlsx)
```
# Read the data and create phyloseq objects
Three tables are needed
* OTU
* Taxonomy
* Samples
## Create analysis-specific phyloseq objects
We maintain **one filtered “base” phyloseq object** and then derive **analysis-specific** objects from it.
This avoids accidental overwriting and ensures each analysis uses the appropriate data scale (counts vs. relative abundance vs. rarefied counts).
- **`ps_raw`** → Raw imported phyloseq object (**integer counts**; import stage only)
- **`ps_base`** → `ps_raw` with **taxonomy + sample metadata** properly aligned (the clean master object before any filtering)
- **`ps_pruned`** → Optional **sample subset** of `ps_base` (e.g., drop unwanted samples by ID/pattern); still **integer counts**
- **`ps_filt`** → The shared filtered backbone: **low-depth samples removed** + taxa with **zero total counts dropped**; remains **integer counts**
```{r, echo=FALSE, warning=FALSE}
library(tidyr)
# For QIIME1
#ps.ng.tax <- import_biom("./exported_table/feature-table.biom", "./exported-tree/tree.nwk")
# For QIIME2
#install.packages("remotes")
#remotes::install_github("jbisanz/qiime2R")
#"core_metrics_results/rarefied_table.qza", rarefying performed in the code, therefore import the raw table.
library(qiime2R)
ps_raw <- qza_to_phyloseq(
features = "dada2_tests/test_59_f235_r245/table.qza",
tree = "rooted-tree.qza",
metadata = "qiime2_metadata_for_qza_to_phyloseq.tsv"
)
# Refresh/ensure sample_data (optional but keeps things explicit)
sample_df <- read.csv("./qiime2_metadata_for_qza_to_phyloseq.tsv", sep="\t", row.names=1, check.names=FALSE)
SAM <- sample_data(sample_df, errorIfNULL = TRUE)
# Add taxonomy table (exported from QIIME2)
taxonomy <- read.delim("./exported-taxonomy/taxonomy.tsv", sep="\t", header=TRUE)
# Separate taxonomy string into separate ranks
taxonomy_df <- taxonomy %>% separate(Taxon, into = c("Domain","Phylum","Class","Order","Family","Genus","Species"), sep = ";", fill = "right", extra = "drop")
# Use Feature.ID as rownames
rownames(taxonomy_df) <- taxonomy_df$Feature.ID
taxonomy_df <- taxonomy_df[, -c(1, ncol(taxonomy_df))] # Drop Feature.ID and Confidence
# Create tax_table
tax_table_final <- phyloseq::tax_table(as.matrix(taxonomy_df))
# Base object: raw integer counts + metadata + taxonomy
ps_base <- merge_phyloseq(ps_raw, SAM, tax_table_final)
print(ps_base)
#colnames(phyloseq::tax_table(ps_base)) <- c("Domain","Phylum","Class","Order","Family","Genus","Species")
saveRDS(ps_base, "./ps_base.rds")
```
Visualize data
```{r, echo=TRUE, warning=FALSE}
# Inspect the base object (raw integer counts)
sample_names(ps_base)
rank_names(ps_base)
sample_variables(ps_base)
# Optional: prune to a naming pattern (avoids hard-coding long sample lists)
#samples_keep <- sample_names(ps_base)
#samples_keep <- samples_keep[grepl("^sample-[A-H]", samples_keep)]
samples_keep <- c("O23092004","O24010402","O23083101","A23080101","PC_8","O23092702","O24010401","O23092001","O23082402","U25011701","O23120702","O23121301","O23112304","U24111801","PC01","A24080201","A23060602","A23051102","UR009768","O24031305","O23090801","UR009909","U24121801","O23120103","U23090801","O23121501","O23110901","A23051103","O24013102","O24011801","O23091403","O23102704","O24020903","A24030402","O23100401","U24101801","O24011105","O24010302","O23121502","O23092005","O24021402","O23102602","NTC_3","O23111502","PC_7","A24071701","O23111501","O24011005","A23112001","U24080201","O24010404","O23112205","O23092803","NTC_6","NTC_8","O24011001","O24010301","O24013104","O24020103","O24011201","O23102601","NTC_2","A23112002","O23102502","O24020901","O24021502","O24021503","O23112901","O23112303","O23112201","O23112902","O24020703","O24010403","O23092003","U23052401","O24021501","O23120701","NTC_11","O24022101","O24011202","O23110902","O23092701","O24011002","A24031201","U23052201","O24021401","O23091303","O24021601","NTC_13","O23092104","O24011102","O23092102","NTC_10","O24022302","A24072901","O24022301","O24022202","O24022901","A24062801","O24011004","O23090602","O23112204","O23100701","O24012403","O24011003","A24071901","O24020802","O24021403","O24021602","NTC_14","NTC_9","PC_1","O24011901","O24013103","O24021404","O24030601","O24020701","O24011204","O24011103","A24030401","NTC_4","O24020902","O23092201","NTC_7","U23071901","O24020904","O23110903","U25011702","U23071701","O24031304","O24030602","O23091501","O24012401","O24030603","O24020704","PC_4","O23110904","O24020804","O23100801","O24030604","O23100802","NTC_15","O24022201","O24012501","O23100706","O23090601","O23100803","O24031301","O23082303","O24010501","O24022902","O23102703","O23110203","O23092202","NTC_12","O23102701","O23082302","O23121304","NTC_16","O24020803","O23090802","O24020101","U25020701","O23121306","O23082301","O23092203","O23110302","O23120101","O24020905","PC_5","O23092802","U25022101","O23100702","PC_6","O23110301","O23081801","O24020705","O24011207","O23091302","O23083001","O23102503","O23121303","O24011206","O24011802","O23102504","O23110204","O24022102","O23120104","O23092002","O23102603","O23092101","O23100503","O23092103","O23111601","O24012402","O24013105","PC_3","O23092801","O23091301","O23100402","O23121503","O24011104","O23112206","O23100703","O23102702","O24022801","O24011203","O23091401","O23090803","O23100705","O23102604","O23091402","PC_2","O23121302","U24070401","O23092006","O23100704","O23112203","O24020702","O24020102","O23112301","O23100804","O24020805","O23121305","O23112202","O24011205","O24013101","O24020801","O23110202","O24031302","O23111602","O23102501","O23110101","O23090701","O24011101","O23091305","O23083102","O24021603","O23112302")
ps_pruned <- prune_samples(samples_keep, ps_base)
# Drop taxa absent after pruning
ps_pruned <- prune_taxa(taxa_sums(ps_pruned) > 0, ps_pruned)
# Quick sanity checks
nsamples(ps_base); ntaxa(ps_base)
nsamples(ps_pruned); ntaxa(ps_pruned)
```
Preprocessing statistics for each sample
```{r, echo=TRUE, warning=FALSE}
denoising_stats <- read.csv("dada2_tests/test_59_f235_r245/data_stats.tsv", sep="\t")
# Display the table
kable(denoising_stats, caption = "Preprocessing statistics for each sample") %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
```
In the QC filtering step, we removed 14 samples that either fell below the minimum sequencing depth (library size < 12,201 reads) or were pre-specified for exclusion via sample_ids (as defined in the previous step). The filtered object (ps_filt) therefore contains only samples meeting the depth cutoff, and taxa were re-pruned to retain only those with nonzero total abundance across the retained samples.
```{r, echo=TRUE, warning=FALSE}
# ------------------------------------------------------------
# Filter low-depth samples (recommended for all analyses)
# ------------------------------------------------------------
min_depth <- 12201 # <-- adjust to your data / study design, keeps all!
ps_filt <- prune_samples(sample_sums(ps_pruned) >= min_depth, ps_pruned)
ps_filt <- prune_taxa(taxa_sums(ps_filt) > 0, ps_filt)
ps_filt
# Keep a depth summary for reporting / QC
depth_summary <- summary(sample_sums(ps_filt))
depth_summary
```
**Differential abundance (DESeq2)** → **`ps_deseq`**: **non-rarefied integer counts** derived from `ps_filt`, with optional **count-based** taxon prefilter
*(default: taxa total counts ≥ 10 across all samples)*
From `ps_filt` (e.g. 5669 taxa and 239 samples), we branch into analysis-ready objects in two directions:
* Direction 1 for diversity analyses
- **Alpha diversity**: `ps_rarefied` ✅ (common)
- **Beta diversity**:
- **Unweighted UniFrac / Jaccard**: `ps_rarefied` ✅ (often recommended)
- **Bray–Curtis / ordination on abundances**: `ps_rel` or Hellinger ✅ (rarefaction optional)
- **Aitchison (CLR)**: CLR-transformed (non-rarefied) ✅ (no rarefaction)
Rarefaction
```{r, echo=TRUE, warning=FALSE}
# RAREFACTION
set.seed(9242) # This will help in reproducing the filtering and nomalisation.
ps_rarefied <- rarefy_even_depth(ps_filt, sample.size = 12201)
# # NORMALIZE number of reads in each sample using median sequencing depth.
# total = median(sample_sums(ps.ng.tax))
# #> total
# #[1] 42369
# standf = function(x, t=total) round(t * (x / sum(x)))
# ps.ng.tax = transform_sample_counts(ps.ng.tax, standf)
# ps_rel <- microbiome::transform(ps.ng.tax, "compositional")
#
# saveRDS(ps.ng.tax, "./ps.ng.tax.rds")
```
* Direction 2 for taxonomic composition plots
- **Taxonomic composition** → **`ps_rel`**: **relative abundance** (compositional) computed **after sample filtering** (e.g. 5669 taxa and 239 samples)
- **Optional cleaner composition plots** → **`ps_abund` / `ps_abund_rel`**: taxa filtered for plotting (e.g., keep taxa with **mean relative abundance > 0.1%**); (e.g. 95 taxa and 239 samples)
`ps_abund` = **counts**, `ps_abund_rel` = **relative abundance** *(use for visualization, not DESeq2)*
For the heatmaps, we focus on the most abundant OTUs by first converting counts to relative abundances within each sample. We then filter to retain only OTUs whose mean relative abundance across all samples exceeds 0.1% (0.001). We are left with 199 OTUs which makes the reading much more easy.
```{r, echo=FALSE, warning=FALSE}
# 1) Convert to relative abundances
ps_rel <- transform_sample_counts(ps_filt, function(x) x / sum(x))
# 2) Get the logical vector of which OTUs to keep (based on relative abundance)
keep_vector <- phyloseq::filter_taxa(
ps_rel,
function(x) mean(x) > 0.001,
prune = FALSE
)
# 3) Use the TRUE/FALSE vector to subset absolute abundance data
ps_abund <- prune_taxa(names(keep_vector)[keep_vector], ps_filt)
# 4) Normalize the final subset to relative abundances per sample
ps_abund_rel <- transform_sample_counts(
ps_abund,
function(x) x / sum(x)
)
```
# Heatmaps
```{r, echo=FALSE, warning=FALSE}
datamat_ = as.data.frame(otu_table(ps_abund))
#datamat <- datamat_[c("1","2","5","6","7", "8","9","10","12","13","14", "15","16","17","18","19","20", "21","22","23","24","25","26","27","28", "29","30","31","32", "33","34","35","36","37","38","39","51", "40","41","42","43","44","46", "47","48","49","50","52","53","55")]
datamat <- datamat_[c("O23092004","O24010402","O23083101","A23080101","PC_8","O23092702","O24010401","O23092001","O23082402","U25011701","O23120702","O23121301","O23112304","U24111801","PC01","A24080201","A23060602","A23051102","UR009768","O24031305","O23090801","UR009909","U24121801","O23120103","U23090801","O23121501","O23110901","A23051103","O24013102","O24011801","O23091403","O23102704","O24020903","A24030402","O23100401","U24101801","O24011105","O24010302","O23121502","O23092005","O24021402","O23102602","NTC_3","O23111502","PC_7","A24071701","O23111501","O24011005","A23112001","U24080201","O24010404","O23112205","O23092803","NTC_6","NTC_8","O24011001","O24010301","O24013104","O24020103","O24011201","O23102601","NTC_2","A23112002","O23102502","O24020901","O24021502","O24021503","O23112901","O23112303","O23112201","O23112902","O24020703","O24010403","O23092003","U23052401","O24021501","O23120701","NTC_11","O24022101","O24011202","O23110902","O23092701","O24011002","A24031201","U23052201","O24021401","O23091303","O24021601","NTC_13","O23092104","O24011102","O23092102","NTC_10","O24022302","A24072901","O24022301","O24022202","O24022901","A24062801","O24011004","O23090602","O23112204","O23100701","O24012403","O24011003","A24071901","O24020802","O24021403","O24021602","NTC_14","NTC_9","PC_1","O24011901","O24013103","O24021404","O24030601","O24020701","O24011204","O24011103","A24030401","NTC_4","O24020902","O23092201","NTC_7","U23071901","O24020904","O23110903","U25011702","U23071701","O24031304","O24030602","O23091501","O24012401","O24030603","O24020704","PC_4","O23110904","O24020804","O23100801","O24030604","O23100802","NTC_15","O24022201","O24012501","O23100706","O23090601","O23100803","O24031301","O23082303","O24010501","O24022902","O23102703","O23110203","O23092202","NTC_12","O23102701","O23082302","O23121304","NTC_16","O24020803","O23090802","O24020101","U25020701","O23121306","O23082301","O23092203","O23110302","O23120101","O24020905","PC_5","O23092802","U25022101","O23100702","PC_6","O23110301","O23081801","O24020705","O24011207","O23091302","O23083001","O23102503","O23121303","O24011206","O24011802","O23102504","O23110204","O24022102","O23120104","O23092002","O23102603","O23092101","O23100503","O23092103","O23111601","O24012402","O24013105","PC_3","O23092801","O23091301","O23100402","O23121503","O24011104","O23112206","O23100703","O23102702","O24022801","O24011203","O23091401","O23090803","O23100705","O23102604","O23091402","PC_2","O23121302","U24070401","O23092006","O23100704","O23112203","O24020702","O24020102","O23112301","O23100804","O24020805","O23121305","O23112202","O24011205","O24013101","O24020801","O23110202","O24031302","O23111602","O23102501","O23110101","O23090701","O24011101","O23091305","O23083102","O24021603","O23112302")]
# Remove rows with zero variance
datamat <- datamat[apply(datamat, 1, var) > 0, ]
# Remove cols with zero variance
#datamat <- datamat[, apply(datamat, 2, var) > 0]
# (optional) replace with your actual column names
#colnames(datamat) <- c(
# "A24040201",...
# )
# ---------- 0) Sample names from datamat ----------
samplenames <- sub("^sample-", "", colnames(datamat))
# ---------- 1) Read metadata ----------
meta_path <- "qiime2_metadata.tsv"
meta <- read.delim(
meta_path,
sep = "\t",
header = TRUE,
stringsAsFactors = FALSE,
check.names = FALSE,
comment.char = ""
)
# ---------- 2) Identify SampleID + Group columns ----------
sample_id_col <- c("#SampleID","SampleID","sample-id","sampleid")
sample_id_col <- sample_id_col[sample_id_col %in% colnames(meta)][1]
if (is.na(sample_id_col)) sample_id_col <- colnames(meta)[1]
group_col <- c("Group","group","GROUP")
group_col <- group_col[group_col %in% colnames(meta)][1]
if (is.na(group_col)) stop("No 'Group' column found in metadata.")
# ---------- 3) Build lookup: sample -> group ----------
meta_ids <- sub("^sample-", "", meta[[sample_id_col]])
meta_groups <- trimws(as.character(meta[[group_col]]))
group_map <- setNames(meta_groups, meta_ids)
# Map datamat columns to group labels
groups <- unname(group_map[samplenames])
groups[is.na(groups)] <- "unknown"
# ---------- 4) Color mapping for YOUR labels ----------
# (Adjust colors if you prefer different ones)
color_map <- c(
"1" = "#a6cee3",
"2" = "#1f78b4",
"3" = "#b2df8a",
"4" = "#33a02c",
"5" = "#fb9a99",
"negative control" = "#6a3d9a",
"positive control" = "#ff7f00",
"unknown" = "GREY"
)
# Assign colors safely
sampleCols <- unname(color_map[groups])
sampleCols[is.na(sampleCols)] <- "GREY"
names(sampleCols) <- samplenames
# ---------- 5) Checks ----------
cat("Unique groups found in datamat:\n")
print(sort(unique(groups)))
cat("\nCounts per group:\n")
print(table(groups, useNA = "ifany"))
cat("\nFirst 10 sample colors:\n")
print(head(sampleCols, 10))
# Optional: list any samples that didn't match metadata
unmatched <- samplenames[groups == "unknown"]
if (length(unmatched) > 0) {
cat("\nWARNING: Unmatched samples (showing up to 20):\n")
print(head(unmatched, 20))
}
```
```{r, echo=TRUE, warning=FALSE, fig.cap="Heatmap", out.width = '100%', fig.align= "center"}
## --- 1) order columns by group (and then by sample name) ---
group_order <- c("1","2","3","4","5","negative control","positive control","unknown")
groups_fac <- factor(groups, levels = group_order, ordered = TRUE)
col_order <- order(groups_fac, samplenames)
datamat_ord <- datamat[, col_order, drop = FALSE]
groups_ord <- groups[col_order]
samplenames_ord <- samplenames[col_order]
sampleCols_ord <- sampleCols[col_order]
stopifnot(identical(colnames(datamat_ord), samplenames_ord))
## group separators
grp_counts <- table(factor(groups_ord, levels = group_order))
grp_breaks <- cumsum(as.integer(grp_counts[grp_counts > 0]))
## --- 2) cluster ROWS using the *ordered* matrix (columns don't matter, but be consistent) ---
hr <- hclust(as.dist(1 - cor(t(datamat_ord), method = "pearson")), method = "complete")
mycl <- cutree(hr, h = max(hr$height) / 1.08)
mycol_palette <- c("YELLOW","DARKBLUE","DARKORANGE","DARKMAGENTA","DARKCYAN","DARKRED",
"MAROON","DARKGREEN","LIGHTBLUE","PINK","MAGENTA","LIGHTCYAN",
"LIGHTGREEN","BLUE","ORANGE","CYAN","RED","GREEN")
mycol <- mycol_palette[as.vector(mycl)]
## --- 3) plot using datamat_ord and sampleCols_ord; keep column order fixed ---
library(RColorBrewer)
heatmap_colors <- colorRampPalette(brewer.pal(9, "Blues"))(100)
png("figures/heatmap.png", width = 1800, height = 2400)
heatmap.2(
as.matrix(datamat_ord),
Rowv = as.dendrogram(hr),
Colv = NA, # IMPORTANT: do NOT cluster columns
dendrogram = "row",
scale = "row",
trace = "none",
col = heatmap_colors,
cexRow = 1.2,
cexCol = 0.8,
RowSideColors = mycol,
ColSideColors = sampleCols_ord, # IMPORTANT: use ordered colors
srtCol = 85,
labRow = row.names(datamat_ord),
labCol = samplenames_ord, # optional but explicit
key = TRUE,
margins = c(10, 15),
lhei = c(0.7, 15),
lwid = c(1, 8),
colsep = grp_breaks, # optional separators
sepcolor = "black",
sepwidth = c(0.02, 0.02)
)
dev.off()
knitr::include_graphics("./figures/heatmap.png")
```
\pagebreak
```{r, echo=FALSE, warning=FALSE}
library(stringr)
#for id in 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199; do
#for id in 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 224 225 226 227 228 229 230 231 232 233 234 235 236 237 238 239 240 241 242 243 244 245 246 247 248 249 250 251 252 253 254 255 256 257 258 259 260 261 262 263 264 265 266 267 268 269 270 271 272 273 274 275 276 277 278 279 280 281 282 283 284 285 286 287 288 289 290 291 292 293 294 295 296 297 298 299 300; do
#for id in 301 302 303 304 305 306 307 308 309 310 311 312 313 314 315 316 317 318 319 320 321 322 323 324 325 326 327 328 329 330 331 332 333 334 335 336 337 338 339 340 341 342 343 344 345 346 347 348 349 350 351 352 353 354 355 356 357 358 359 360 361 362 363 364 365 366 367 368 369 370 371 372 373 374 375 376 377 378 379 380 381 382; do
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Domain\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Domain\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Phylum\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Phylum\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Class\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Class\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Order\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Order\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Family\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Family\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Genus\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Genus\"], \"__\")[[1]][2]"
# echo "phyloseq::tax_table(ps_abund_rel)[${id},\"Species\"] <- str_split(phyloseq::tax_table(ps_abund_rel)[${id},\"Species\"], \"__\")[[1]][2]"
#done
phyloseq::tax_table(ps_abund_rel)[1,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[1,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[1,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[2,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[2,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[3,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[3,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[4,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[4,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[5,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[5,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[6,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[6,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[7,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[7,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[8,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[8,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[9,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[9,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[10,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[10,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[11,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[11,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[12,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[12,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[13,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[13,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[14,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[14,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[15,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[15,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[16,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[16,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[17,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[17,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[18,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[18,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[19,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[19,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[20,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[20,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[21,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[21,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[22,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[22,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[23,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[23,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[24,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[24,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[25,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[25,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[26,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[26,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[27,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[27,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[28,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[28,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[29,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[29,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[30,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[30,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[31,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[31,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[32,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[32,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[33,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[33,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[34,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[34,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[35,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[35,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[36,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[36,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[37,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[37,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[38,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[38,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[39,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[39,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[40,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[40,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[41,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[41,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[42,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[42,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[43,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[43,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[44,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[44,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[45,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[45,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[46,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[46,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[47,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[47,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[48,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[48,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[49,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[49,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[50,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[50,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[51,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[51,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[52,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[52,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[53,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[53,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[54,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[54,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[55,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[55,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[56,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[56,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[57,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[57,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[58,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[58,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[59,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[59,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[60,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[60,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[61,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[61,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[62,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[62,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[63,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[63,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[64,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[64,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[65,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[65,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[66,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[66,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[67,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[67,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[68,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[68,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[69,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[69,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[70,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[70,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[71,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[71,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[72,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[72,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[73,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[73,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[74,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[74,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[75,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[75,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[76,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[76,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[77,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[77,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[78,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[78,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[79,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[79,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[80,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[80,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[81,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[81,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[82,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[82,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[83,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[83,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[84,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[84,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[85,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[85,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[86,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[86,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[87,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[87,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[88,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[88,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[89,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[89,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[90,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[90,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[91,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[91,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[92,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[92,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[93,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[93,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[94,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[94,"Species"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Domain"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Domain"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Phylum"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Phylum"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Class"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Class"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Order"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Order"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Family"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Family"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Genus"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Genus"], "__")[[1]][2]
phyloseq::tax_table(ps_abund_rel)[95,"Species"] <- str_split(phyloseq::tax_table(ps_abund_rel)[95,"Species"], "__")[[1]][2]
```
# Taxonomic summary
## Bar plots in phylum level
```{r, fig.width=16, fig.height=8, echo=TRUE, warning=FALSE}
sample_order <- c(
"U24080201","U25020701","O23092004","U24101801","U25022101","O23102703","A24062801","O23112205","U23071901","A23112002","U24111801","O23110101","U24121801","O23120101","O24011202","O23090803","A23060602","A24030402","U25011701","O24011901","U23090801","O24011201","O24011003","O23092202","O23082301","O23091403","O23112901","O23092201","O24013103","O24021403","O24010402","O23092005","O23092203","O24010302","O23090701","O23091501","O23092701","O24022202","O23092802","O23090601","O23100401","O24022102","O23081801","O23092006","O23100503","O23090602","O24013104","O24020803","O24010301","O24010404","O23090802","O23092801","O24022801","O23100706","O23102602","O24021601","O24012401","O24021603","O24022901","O24021501","O23110902","O23102601","O23102704","O23100803","O23102701","O24021401","O24022101","O24030603","O23110901","O23110903","O23110301","O24022301","O23102502","O23111501","O23111602","O24020705","O24021502","O24022201","O23110202","O23090801","U23071701","U23052401","U23052201","U24070401","O24011801","O23092003","A24071901","A24072901","O24011102","O23121501","O23092104","O23092001","O23121301","O24020701","O23112201","O23100701","O23100801","O24020903","O24020901","O24020703","O23112204","O23100802","O24011205","O23092002","O24011207","O23092103","O23102501","O24011005","A24030401","O24011004","A23051102","U25011702","O24011204","O23121502","O23120702","O24011206","O24021404","O23092101","O24010403","O23112303","O23083001","O23082302","O24010401","O24022302","O24010501","O23112902","O23082303","O23083102","O24013101","O23100402","O24020801","O23120701","O23121304","O24021602","O24011802","O23121306","O23120103","O24020905","O24012403","O24013102","O24021503","O24020904","O23102504","O24013105","O24030601","O23100705","O24030604","O23111601","O24020103","O24030602","O23110302","O23102603","O24031304","O24021402","O24020101","O24012501","O24020804","O23100804","O23102503","O24022902","O24020704","O23110904","O24020102","O24012402","O23102702","O23102604","O23110204","O23110203","O23083101","O23092702","O23112304","A23051103","A24071701","A23080101","A24031201","A24080201","O24011105","O23091305","O23121302","O23092803","O23091303","O23112301","O24011203","A23112001","O24011001","O24011002","O23091302","O24020902","O23092102","O23091401","O23121503","O23091402","O24020702","O23091301","O23112206","O24011103","O23121305","O24011101","O23121303","O24011104","O23120104","O23100704","O23112302","O23112203","O23100703","O24020805","O24020802","O23112202","O24031302","O23111502","O23100702","O24031301","O24031305","O23082402","NTC_2","NTC_3","NTC_16","NTC_4","NTC_6","NTC_7","NTC_8","NTC_9","NTC_11","NTC_12","NTC_13","NTC_14","NTC_15","NTC_10","UR009768","UR009909","PC01","PC_1","PC_2","PC_8","PC_4","PC_3","PC_5","PC_6","PC_7"
)
# create a sample ID column in sample_data (or overwrite an existing one)
sample_data(ps_abund_rel)$SampleID <- sample_names(ps_abund_rel)
# set the order as a factor with the desired levels
sample_data(ps_abund_rel)$SampleID <- factor(
sample_data(ps_abund_rel)$SampleID,
levels = sample_order
)
#aes(color="Phylum", fill="Phylum") --> aes()
#ggplot(data=data, aes(x=Sample, y=Abundance, fill=Phylum))
my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, x = "SampleID", fill = "Phylum") +
geom_bar(stat = "identity", position = "stack") +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(
angle = 85, # 85° rotation
hjust = 1, # right-justified so labels are neat
vjust = 1,
size = 5,
colour = "black"
),
axis.text.y = element_text(size = 7, colour = "black"),
legend.position = "bottom"
) +
guides(fill = guide_legend(nrow = 2))
```
### Aggregate samples by group and normalize read counts within each group to correct for differences in sequencing depth.
```{r, echo=TRUE, warning=FALSE}
# merge + normalize
ps_abund_rel_group <- merge_samples(ps_abund_rel, "Group")
ps_abund_rel_group_ <- transform_sample_counts(
ps_abund_rel_group,
function(x) x / sum(x)
)
# desired order on x-axis
group_order <- c("1", "2", "3", "4", "5", "negative control", "positive control")
plot_bar(ps_abund_rel_group_, fill = "Phylum") +
geom_bar(stat = "identity", position = "stack") +
scale_x_discrete(limits = group_order) + # <-- set order here
scale_fill_manual(values = my_colors) +
labs(x = "Group") + # <- change x-axis label from "Sample" to "Group"
theme(axis.text = element_text(angle = 0, size = 10, colour="black"), axis.text.x = element_text(angle = -15),hjust = 10,vjust = 2)
```
## Bar plots in class level
```{r, fig.width=16, fig.height=8, echo=TRUE, warning=FALSE}
my_colors <- c("darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "lightskyblue", "darkgreen", "deeppink", "khaki2", "firebrick", "brown1", "darkorange1", "cyan1", "royalblue4", "darksalmon", "darkblue","royalblue4", "dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen", "darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1", "brown1", "darkorange1", "cyan1", "darkgrey")
plot_bar(ps_abund_rel, x = "SampleID", fill = "Class") +
geom_bar(stat = "identity", position = "stack") +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(
angle = 85,
hjust = 1,
vjust = 1,
size = 5,
colour = "black"
),
axis.text.y = element_text(size = 7, colour = "black"),
legend.position = "bottom"
) +
guides(fill = guide_legend(nrow = 3))
```
### Aggregate samples by group and normalize read counts within each group to correct for differences in sequencing depth.
```{r, echo=TRUE, warning=FALSE}
plot_bar(ps_abund_rel_group_, fill="Class") + geom_bar(aes(), stat="identity", position="stack") + scale_x_discrete(limits = group_order) +
scale_fill_manual(values = my_colors) + labs(x = "Group") + theme(axis.text = element_text(angle = 0, size = 10, colour="black"), axis.text.x = element_text(angle = -15),hjust = 10,vjust = 2)
```
\pagebreak
## Bar plots in order level
```{r, fig.width=16, fig.height=8, echo=TRUE, warning=FALSE}
my_colors <- c(
"darkblue", "darkgoldenrod1", "darkseagreen", "darkorchid",
"darkolivegreen1", "lightskyblue", "darkgreen", "deeppink",
"khaki2", "firebrick", "brown1", "darkorange1", "cyan1",
"royalblue4", "darksalmon", "darkblue","royalblue4",
"dodgerblue3", "steelblue1", "lightskyblue", "darkseagreen",
"darkgoldenrod1", "darkseagreen", "darkorchid", "darkolivegreen1",
"brown1", "darkorange1", "cyan1", "darkgrey"
)
plot_bar(ps_abund_rel, x = "SampleID", fill = "Order") +
geom_bar(stat = "identity", position = "stack") +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(
angle = 85,
hjust = 1,
vjust = 1,
size = 5,
colour = "black"
),
axis.text.y = element_text(size = 7, colour = "black"),
legend.position = "bottom"
) +
guides(fill = guide_legend(nrow = 4))
```
### Aggregate samples by group and normalize read counts within each group to correct for differences in sequencing depth.
```{r, echo=TRUE, warning=FALSE}
plot_bar(ps_abund_rel_group_, fill="Order") + geom_bar(aes(), stat="identity", position="stack") + scale_x_discrete(limits = group_order) +
scale_fill_manual(values = my_colors) + labs(x = "Group") + theme(axis.text = element_text(angle = 0, size = 10, colour="black"), axis.text.x = element_text(angle = -15),hjust = 10,vjust = 2)
```
\pagebreak
## Bar plots in family level
```{r, fig.width=16, fig.height=8, echo=TRUE, warning=FALSE}
my_colors <- c(
"#FF0000", "#000000", "#0000FF", "#C0C0C0", "#FFFFFF", "#FFFF00", "#00FFFF", "#FFA500", "#00FF00", "#808080", "#FF00FF", "#800080", "#FDD017", "#0000A0", "#3BB9FF", "#008000", "#800000", "#ADD8E6", "#F778A1", "#800517", "#736F6E", "#F52887", "#C11B17", "#5CB3FF", "#A52A2A", "#FF8040", "#2B60DE", "#736AFF", "#1589FF", "#98AFC7", "#8D38C9", "#307D7E", "#F6358A", "#151B54", "#6D7B8D", "#FDEEF4", "#FF0080", "#F88017", "#2554C7", "#FFF8C6", "#D4A017", "#306EFF", "#151B8D", "#9E7BFF", "#EAC117", "#E0FFFF", "#15317E", "#6C2DC7", "#FBB917", "#FCDFFF", "#15317E", "#254117", "#FAAFBE", "#357EC7"
)
plot_bar(ps_abund_rel, x = "SampleID", fill = "Family") +
geom_bar(stat = "identity", position = "stack") +
scale_fill_manual(values = my_colors) +
theme(
axis.text.x = element_text(
angle = 85,
hjust = 1,
vjust = 1,
size = 5,
colour = "black"
),
axis.text.y = element_text(size = 7, colour = "black"),
legend.position = "bottom"
) +
guides(fill = guide_legend(nrow = 8))
```
### Aggregate samples by group and normalize read counts within each group to correct for differences in sequencing depth.
```{r, echo=TRUE, warning=FALSE}
plot_bar(ps_abund_rel_group_, fill="Family") + geom_bar(aes(), stat="identity", position="stack") + scale_x_discrete(limits = group_order) +
scale_fill_manual(values = my_colors) + labs(x = "Group") + theme(axis.text = element_text(angle = 0, size = 10, colour="black"), axis.text.x = element_text(angle = -15),hjust = 10,vjust = 2)
```
\pagebreak
```{r, fig.width=16, fig.height=8, echo=FALSE, warning=FALSE}
# !!!!NOT_USED!!!!: #Export Relative abundances of Phylum, Class, Order, and Family levels across all samples in Excel files!
library(phyloseq)
library(writexl)
library(dplyr)
# Function to check for NA or empty values in a taxonomic rank
check_taxa_names <- function(tax_table, rank) {
tax_values <- tax_table[[rank]]
na_count <- sum(is.na(tax_values) | tax_values == "")
cat("Number of NA or empty values in", rank, ":", na_count, "\n")
if (na_count > 0) {
cat("Taxa with NA or empty", rank, ":\n")
print(tax_values[is.na(tax_values) | tax_values == ""])
}
}
# Function to create and save relative abundance table for a given taxonomic rank with normalization
save_taxa_abundance <- function(ps, rank, output_file) {
# Check for NA or empty values in the taxonomy table
tax_table_df <- as.data.frame(tax_table(ps))
check_taxa_names(tax_table_df, rank)
# Aggregate OTUs by taxonomic rank, removing taxa with NA at the specified rank
ps_glom <- tax_glom(ps, taxrank = rank, NArm = TRUE)
# Extract OTU table (relative abundances)
otu_table <- as.data.frame(otu_table(ps_glom))
# Normalize each column to sum to 1
otu_table_normalized <- apply(otu_table, 2, function(x) x / sum(x))
# Convert matrix to data frame
otu_table_normalized <- as.data.frame(otu_table_normalized)
# Verify column sums are approximately 1.0
col_sums <- colSums(otu_table_normalized)
if (any(abs(col_sums - 1) > 1e-6)) {
warning("Column sums in ", rank, " table do not equal 1.0: ", paste(col_sums, collapse = ", "))
} else {
cat("Column sums for ", rank, " table are all approximately 1.0\n")
}
# Extract taxonomy table and get the specified rank for taxa names
tax_table_glom <- as.data.frame(tax_table(ps_glom))
taxa_names <- tax_table_glom[[rank]]
# Replace NA or empty strings with "Unclassified"
taxa_names <- ifelse(is.na(taxa_names) | taxa_names == "", paste0("Unclassified_", rank), taxa_names)
# Ensure unique row names
taxa_names <- make.unique(taxa_names)
# Set row names to taxa names (for internal reference)
rownames(otu_table_normalized) <- taxa_names
# Add taxa names as a column
otu_table_normalized[[rank]] <- taxa_names
# Reorder to move rank column to the first position
otu_table_normalized <- otu_table_normalized[, c(rank, setdiff(names(otu_table_normalized), rank))]
# Rename sample columns by removing "sample-" prefix
names(otu_table_normalized)[-1] <- sub("sample-", "", names(otu_table_normalized)[-1])
# Write the data frame to Excel, including the rank column
write_xlsx(otu_table_normalized, path = output_file)
cat("Saved", output_file, "\n")
}
# Verify column sums of ps_abund_rel
col_sums <- colSums(otu_table(ps_abund_rel))
cat("Column sums of ps_abund_rel:\n")
summary(col_sums)
## Generate Excel files for Phylum, Class, Order, and Family levels with normalization and renamed sample names
#save_taxa_abundance(ps_abund_rel, "Phylum", "relative_abundance_phylum_old.xlsx")
#save_taxa_abundance(ps_abund_rel, "Class", "relative_abundance_class_old.xlsx")
#save_taxa_abundance(ps_abund_rel, "Order", "relative_abundance_order_old.xlsx")
#save_taxa_abundance(ps_abund_rel, "Family", "relative_abundance_family_old.xlsx")
```
```{r, fig.width=16, fig.height=8, echo=FALSE, warning=FALSE}
library(phyloseq)
library(writexl)
library(dplyr)
# Function to check for NA or empty values in a taxonomic rank
check_taxa_names <- function(tax_table, rank) {
tax_values <- tax_table[[rank]]
na_count <- sum(is.na(tax_values) | tax_values == "")
cat("Number of NA or empty values in", rank, ":", na_count, "\n")
if (na_count > 0) {
cat("Taxa with NA or empty", rank, ":\n")
print(tax_values[is.na(tax_values) | tax_values == ""])
}
}
# Function to create and save relative abundance table for a given taxonomic rank with normalization
save_taxa_abundance <- function(ps, rank, output_file) {
# Clean the taxonomy table by removing D_[level]__ prefixes
tax_table_df <- as.data.frame(tax_table(ps))
tax_table_df[[rank]] <- ifelse(is.na(tax_table_df[[rank]]) | tax_table_df[[rank]] == "",
paste0("Unclassified_", rank),
sub("^D_[0-9]+__(.+)", "\\1", tax_table_df[[rank]]))
tax_table(ps) <- as.matrix(tax_table_df) # Update taxonomy table with cleaned names
# Check for NA or empty values in the taxonomy table
check_taxa_names(tax_table_df, rank)
# Aggregate OTUs by taxonomic rank, removing taxa with NA at the specified rank
ps_glom <- tax_glom(ps, taxrank = rank, NArm = TRUE)
# Extract OTU table (relative abundances)
otu_table <- as.data.frame(otu_table(ps_glom))
# Normalize each column to sum to 1
otu_table_normalized <- apply(otu_table, 2, function(x) x / sum(x))
# Convert matrix to data frame
otu_table_normalized <- as.data.frame(otu_table_normalized)
# Verify column sums are approximately 1.0
col_sums <- colSums(otu_table_normalized)
if (any(abs(col_sums - 1) > 1e-6)) {
warning("Column sums in ", rank, " table do not equal 1.0: ", paste(col_sums, collapse = ", "))
} else {
cat("Column sums for ", rank, " table are all approximately 1.0\n")
}
# Extract taxonomy table and get the specified rank for taxa names
tax_table_glom <- as.data.frame(tax_table(ps_glom))
taxa_names <- tax_table_glom[[rank]]
# Ensure unique row names
taxa_names <- make.unique(taxa_names)
# Set row names to taxa names (for internal reference)
rownames(otu_table_normalized) <- taxa_names
# Add taxa names as a column
otu_table_normalized[[rank]] <- taxa_names
# Reorder to move rank column to the first position
otu_table_normalized <- otu_table_normalized[, c(rank, setdiff(names(otu_table_normalized), rank))]
# Rename sample columns by removing "sample-" prefix
names(otu_table_normalized)[-1] <- sub("sample-", "", names(otu_table_normalized)[-1])
# Write the data frame to Excel, including the rank column
write_xlsx(otu_table_normalized, path = output_file)
cat("Saved", output_file, "\n")
}
# Verify column sums of ps_abund_rel
col_sums <- colSums(otu_table(ps_abund_rel))
cat("Column sums of ps_abund_rel:\n")
summary(col_sums)
## Generate Excel files for Phylum, Class, Order, and Family levels with normalization and renamed sample names
#save_taxa_abundance(ps_abund_rel, "Phylum", "relative_abundance_phylum.xlsx")
#save_taxa_abundance(ps_abund_rel, "Class", "relative_abundance_class.xlsx")
#save_taxa_abundance(ps_abund_rel, "Order", "relative_abundance_order.xlsx")
#save_taxa_abundance(ps_abund_rel, "Family", "relative_abundance_family.xlsx")
#Sum up the last two colums with the same row.names to a new column, export the file as csv, then delete the two rows before last, then merge them with csv2xls to a Excel-file, adapt the sheet-names.
#~/Tools/csv2xls-0.4/csv_to_xls.py relative_abundance_phylum.csv relative_abundance_order.csv relative_abundance_family.csv -d$'\t' -o relative_abundance_phylum_order_family.xls;
```
\pagebreak
# Alpha diversity
Plot Chao1 richness estimator, Observed OTUs, Shannon index, and Phylogenetic diversity.
Regroup together samples from the same group.
```{r, echo=FALSE, warning=FALSE}
# using rarefied data
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even42369.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering/rep_set.tre
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even4753.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering_stool/rep_set.tre
#gunzip table_even4753.biom.gz
#alpha_diversity.py -i table_even4753.biom --metrics chao1,observed_otus,shannon,PD_whole_tree -o adiv_even.txt -t ../clustering_swab/rep_set.tre
```
```{r, echo=TRUE, warning=FALSE}
hmp.meta <- meta(ps_rarefied)
hmp.meta$sam_name <- rownames(hmp.meta)
# ---- enforce Group order (edit if you have different labels) ----
group_order <- c("1","2","3","4","5","negative control", "positive control")
hmp.meta$Group <- factor(as.character(hmp.meta$Group), levels = group_order)
# for QIIME2: Lesen der Metriken
shannon <- read.table("exported_alpha/shannon/alpha-diversity.tsv", header=TRUE, sep="\t") #cp -r ../Data_Karoline_16S_2025/exported_alpha/ .
faith_pd <- read.table("exported_alpha/faith_pd/alpha-diversity.tsv", header=TRUE, sep="\t")
observed <- read.table("exported_alpha/observed_features/alpha-diversity.tsv", header=TRUE, sep="\t")
#chao1 <- read.table("exported_alpha/chao1/alpha-diversity.tsv", header=TRUE, sep="\t") #TODO: Check the correctness of chao1-calculation.
# Umbenennen für Klarheit
colnames(shannon) <- c("sam_name", "shannon")
colnames(faith_pd) <- c("sam_name", "PD_whole_tree")
colnames(observed) <- c("sam_name", "observed_otus")
#colnames(chao1) <- c("sam_name", "chao1")
# Merge alles in ein DataFrame
div.df <- Reduce(function(x, y) merge(x, y, by="sam_name"),
list(shannon, faith_pd, observed))
# Meta-Daten einfügen
div.df <- merge(div.df, hmp.meta, by="sam_name")
# Reformat
div.df2 <- div.df[, c("sam_name", "Group", "shannon", "observed_otus", "PD_whole_tree")]
colnames(div.df2) <- c("Sample name", "Group", "Shannon", "OTU", "Phylogenetic Diversity")
write.csv(div.df2, file="alpha_diversities.txt")
knitr::kable(div.df2) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
#https://uc-r.github.io/t_test
#We can perform the test with t.test and transform our data and we can also perform the nonparametric test with the wilcox.test function.
stat.test.Shannon <- compare_means(
Shannon ~ Group, data = div.df2,
method = "t.test"
)
knitr::kable(stat.test.Shannon) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
div_df_melt <- reshape2::melt(div.df2)
#head(div_df_melt)
#https://plot.ly/r/box-plots/#horizontal-boxplot
#http://www.sthda.com/english/wiki/print.php?id=177
#https://rpkgs.datanovia.com/ggpubr/reference/as_ggplot.html
#http://www.sthda.com/english/articles/24-ggpubr-publication-ready-plots/82-ggplot2-easy-way-to-change-graphical-parameters/
#https://plot.ly/r/box-plots/#horizontal-boxplot
#library("gridExtra")
#par(mfrow=c(4,1))
p <- ggboxplot(div_df_melt, x = "Group", y = "value",
facet.by = "variable",
scales = "free",
width = 0.5,
fill = "gray", legend= "right")
#ggpar(p, xlab = FALSE, ylab = FALSE)
lev <- levels(factor(div_df_melt$Group)) # get the variables
#FITTING4: delete H47(1) in lev
#lev <- lev[-c(3)]
# make a pairwise list that we want to compare.
#my_stat_compare_means
#https://stackoverflow.com/questions/47839988/indicating-significance-with-ggplot2-in-a-boxplot-with-multiple-groups
L.pairs <- combn(seq_along(lev), 2, simplify = FALSE, FUN = function(i) lev[i]) #%>% filter(p.signif != "ns")
my_stat_compare_means <- function (mapping = NULL, data = NULL, method = NULL, paired = FALSE,
method.args = list(), ref.group = NULL, comparisons = NULL,
hide.ns = FALSE, label.sep = ", ", label = NULL, label.x.npc = "left",
label.y.npc = "top", label.x = NULL, label.y = NULL, tip.length = 0.03,
symnum.args = list(), geom = "text", position = "identity",
na.rm = FALSE, show.legend = NA, inherit.aes = TRUE, ...)
{
if (!is.null(comparisons)) {
method.info <- ggpubr:::.method_info(method)
method <- method.info$method
method.args <- ggpubr:::.add_item(method.args, paired = paired)
if (method == "wilcox.test")
method.args$exact <- FALSE
pms <- list(...)
size <- ifelse(is.null(pms$size), 0.3, pms$size)
color <- ifelse(is.null(pms$color), "black", pms$color)
map_signif_level <- FALSE
if (is.null(label))
label <- "p.format"
if (ggpubr:::.is_p.signif_in_mapping(mapping) | (label %in% "p.signif")) {
if (ggpubr:::.is_empty(symnum.args)) {
map_signif_level <- c(`****` = 1e-04, `***` = 0.001,
`**` = 0.01, `*` = 0.05, ns = 1)
} else {
map_signif_level <- symnum.args
}
if (hide.ns)
names(map_signif_level)[5] <- " "
}
step_increase <- ifelse(is.null(label.y), 0.12, 0)
ggsignif::geom_signif(comparisons = comparisons, y_position = label.y,
test = method, test.args = method.args, step_increase = step_increase,
size = size, color = color, map_signif_level = map_signif_level,
tip_length = tip.length, data = data)
} else {
mapping <- ggpubr:::.update_mapping(mapping, label)
layer(stat = StatCompareMeans, data = data, mapping = mapping,
geom = geom, position = position, show.legend = show.legend,
inherit.aes = inherit.aes, params = list(label.x.npc = label.x.npc,
label.y.npc = label.y.npc, label.x = label.x,
label.y = label.y, label.sep = label.sep, method = method,
method.args = method.args, paired = paired, ref.group = ref.group,
symnum.args = symnum.args, hide.ns = hide.ns,
na.rm = na.rm, ...))
}
}
# Rotate the x-axis labels to 45 degrees and adjust their position
p <- p + theme(axis.text.x = element_text(angle = 45, hjust = 1, vjust=1, size=8))
p2 <- p +
stat_compare_means(
method="t.test",
comparisons = list(c("1", "2"), c("1", "3"), c("1", "4"), c("1", "5"), c("2", "3"), c("2", "4"), c("2", "5"), c("3", "4"), c("3", "5"), c("4", "5")),
label = "p.signif",
symnum.args <- list(cutpoints = c(0, 0.0001, 0.001, 0.01, 0.05, 1), symbols = c("****", "***", "**", "*", "ns"))
)
#comparisons = L.pairs,
#symnum.args <- list(cutpoints = c(0, 0.0001, 0.001, 0.01, 0.05), symbols = c("****", "***", "**", "*")),
#stat_pvalue_manual
print(p2)
#https://stackoverflow.com/questions/20500706/saving-multiple-ggplots-from-ls-into-one-and-separate-files-in-r
ggsave("./figures/alpha_diversity_Group.png", device="png", height = 10, width = 15)
ggsave("./figures/alpha_diversity_Group.svg", device="svg", height = 10, width = 15)
```
```{r, echo=FALSE, warning=FALSE, fig.cap="Alpha diversity", out.width = '100%', fig.align= "center"}
## MANUALLY selected alpha diversities unter host-env after 'cp alpha_diversities.txt selected_alpha_diversities.txt'
#knitr::include_graphics("./figures/alpha_diversity_Group.png")
#selected_alpha_diversities<-read.csv("selected_alpha_diversities.txt",sep="\t")
#knitr::kable(selected_alpha_diversities) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
```
```{r, echo=FALSE, warning=FALSE, out.width = '100%', fig.align= "center"}
#!!# Beta diversity (Bray-Curtis distance)
#!!## Group1 vs Group2
#fig.cap="Beta diversity",
#for QIIME1: file:///home/jhuang/DATA/Data_Marius_16S/core_diversity_e42369/bdiv_even42369_Group/weighted_unifrac_boxplots/Group_Stats.txt
# -- for QIIME2: MANUALLY filter permanova-pairwise.csv and save as permanova-pairwise_.csv
# #grep "Permutations" exported_beta_group/permanova-pairwise.csv > permanova-pairwise_.csv
# #grep "Group1,Group2" exported_beta_group/permanova-pairwise.csv >> permanova-pairwise_.csv
# #grep "Group3,Group4" exported_beta_group/permanova-pairwise.csv >> permanova-pairwise_.csv
# beta_diversity_group_stats<-read.csv("permanova-pairwise_.csv",sep=",")
# #beta_diversity_group_stats <- beta_diversity_group_stats[beta_diversity_group_stats$Group.1 == "Group1" & beta_diversity_group_stats$Group.2 == "Group2", ]
# #beta_diversity_group_stats <- beta_diversity_group_stats[beta_diversity_group_stats$Group.1 == "Group3" & beta_diversity_group_stats$Group.2 == "Group4", ]
# knitr::kable(beta_diversity_group_stats) %>% kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
#NOTE: Run this Phyloseq.Rmd, then run the code of MicrobiotaProcess.R to manually generate Comparison_of_Bray_Distances_Group1_vs_Group2.png and Comparison_of_Bray_Distances_Group3_vs_Group4.png, then run this Phyloseq.Rmd!
#knitr::include_graphics("./figures/Comparison_of_Bray_Distances_Group1_vs_Group2.png")
```
# Principal coordinates analysis (PCoA) based on Bray–Curtis dissimilarity
Global PERMANOVA on the weighted UniFrac distance matrix indicated a significant effect of Group on overall community composition (adonis2: R² = 0.1606, F = 7.397, p = 1×10⁻⁴; 9,999 permutations).
```{r, echo=FALSE, results='asis'}
# --- Global beta-diversity (PERMANOVA) ---
cat("```text\n")
cat(
"[PERMANOVA result]\n",
"The object contained internal attribute: PCoA ADONIS\n",
"Permutation test for adonis under reduced model\n",
"Permutation: free\n",
"Number of permutations: 9999\n\n",
"vegan::adonis2(formula = .formula, data = sampleda, permutations = permutations, method = distmethod)\n",
" Df SumOfSqs R2 F Pr(>F)\n",
"Model 6 11.446 0.16058 7.3971 1e-04 ***\n",
"Residual 232 59.829 0.83942\n",
"Total 238 71.274 1.00000\n",
"---\n",
"Signif. codes: 0 ‘***’ 0.001 ‘**’ 0.01 ‘*’ 0.05 ‘.’ 0.1 ‘ ’ 1\n",
sep = ""
)
cat("```\n")
```
Pairwise PERMANOVA tests were performed on Bray–Curtis distance matrices to compare bacterial community composition between all pairs of sample groups (metadata column Group). For each pairwise comparison, the distance matrix was subset to samples from the two groups only, and significance was assessed using vegan::adonis2 with 9,999 permutations. Resulting p-values were adjusted for multiple testing using both Benjamini–Hochberg (BH/FDR) and Bonferroni corrections.
```{r, echo=FALSE, warning=FALSE, out.width = '100%', fig.align= "center"}
#, and the full results were exported to figures/Bray_pairwise_PERMANOVA.csv
# --- Pairwise PERMANOVA results ---
Bray_pairwise_PERMANOVA <- read.csv("figures/Bray_pairwise_PERMANOVA.csv", sep = ",")
knitr::kable(Bray_pairwise_PERMANOVA, caption = "Pairwise PERMANOVA results (distance-based community differences among Group levels).") %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
# --- Ordination figures ---
#knitr::include_graphics("./PCoA.png")
knitr::include_graphics("./PCoA2.png")
knitr::include_graphics("./PCoA3.png")
```
# Differential abundance analysis
Differential abundance analysis aims to find the differences in the abundance of each taxa between two groups of samples, assigning a significance value to each comparison.
```{r, echo=FALSE, warning=FALSE}
# ------------------------------------------------------------
# DESeq2: non-rarefied integer counts + optional taxon prefilter
# ------------------------------------------------------------
ps_deseq <- ps_filt
Group1<-c("U24080201","U25020701","O23092004","U24101801","U25022101","O23102703","A24062801","O23112205","U23071901","A23112002","U24111801","O23110101","U24121801","O23120101","O24011202","O23090803","A23060602","A24030402","U25011701","O24011901","U23090801","O24011201","O24011003","O23092202","O23082301","O23091403","O23112901","O23092201","O24013103","O24021403","O24010402","O23092005","O23092203","O24010302","O23090701","O23091501","O23092701","O24022202","O23092802","O23090601","O23100401","O24022102","O23081801","O23092006","O23100503","O23090602","O24013104","O24020803","O24010301","O24010404","O23090802","O23092801","O24022801","O23100706","O23102602","O24021601","O24012401","O24021603","O24022901","O24021501","O23110902","O23102601","O23102704","O23100803","O23102701","O24021401","O24022101","O24030603","O23110901","O23110903","O23110301","O24022301","O23102502","O23111501","O23111602","O24020705","O24021502","O24022201","O23110202","O23090801")
Group2<-c("U23071701","U23052401","U23052201","U24070401","O24011801","O23092003","A24071901","A24072901","O24011102","O23121501","O23092104","O23092001","O23121301","O24020701","O23112201","O23100701","O23100801","O24020903","O24020901","O24020703","O23112204")
Group3<-c("O23100802","O24011205","O23092002","O24011207","O23092103","O23102501","O24011005","A24030401","O24011004","A23051102","U25011702","O24011204","O23121502","O23120702","O24011206","O24021404","O23092101","O24010403","O23112303","O23083001","O23082302","O24010401","O24022302","O24010501","O23112902","O23082303","O23083102","O24013101","O23100402","O24020801","O23120701","O23121304","O24021602","O24011802","O23121306","O23120103","O24020905","O24012403","O24013102","O24021503","O24020904","O23102504","O24013105","O24030601","O23100705","O24030604","O23111601","O24020103","O24030602","O23110302","O23102603","O24031304","O24021402","O24020101","O24012501","O24020804","O23100804","O23102503","O24022902","O24020704","O23110904","O24020102","O24012402","O23102702","O23102604","O23110204","O23110203","O23083101","O23092702")
Group4<-c("O23112304","A23051103","A24071701","A23080101","A24031201","A24080201","O24011105","O23091305","O23121302","O23092803")
Group5<-c("O23091303","O23112301","O24011203","A23112001","O24011001","O24011002","O23091302","O24020902","O23092102","O23091401","O23121503","O23091402","O24020702","O23091301","O23112206","O24011103","O23121305","O24011101","O23121303","O24011104","O23120104","O23100704","O23112302","O23112203","O23100703","O24020805","O24020802","O23112202","O24031302","O23111502","O23100702","O24031301","O24031305","O23082402")
NTC<-c("NTC_2","NTC_3","NTC_16","NTC_4","NTC_6","NTC_7","NTC_8","NTC_9","NTC_11","NTC_12","NTC_13","NTC_14","NTC_15","NTC_10")
PC<-c("PC_1","PC_2","PC_8","PC_4","PC_3","PC_5","PC_6","PC_7","UR009768","UR009909","PC01")
```
## Group 1 vs Group 2
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group1,Group2)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "2")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group1_vs_Group2"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 1 vs Group 3
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group1,Group3)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "3")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group1_vs_Group3"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 1 vs Group 4
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group1,Group4)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "4")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group1_vs_Group4"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 1 vs Group 5
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group1,Group5)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "5")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group1_vs_Group5"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 2 vs Group 3
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group2,Group3)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "3")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group2_vs_Group3"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 2 vs Group 4
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group2,Group4)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "4")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group2_vs_Group4"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 2 vs Group 5
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group2,Group5)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "5")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group2_vs_Group5"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 3 vs Group 4
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group3,Group4)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "4")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group3_vs_Group4"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 3 vs Group 5
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group3,Group5)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "5")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group3_vs_Group5"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```
## Group 4 vs Group 5
```{r, echo=TRUE, warning=FALSE}
ps_deseq_sel <- data.table::copy(ps_deseq)
otu_table(ps_deseq_sel) <- otu_table(ps_deseq)[,c(Group4,Group5)]
diagdds = phyloseq_to_deseq2(ps_deseq_sel, ~Group)
diagdds$Group <- relevel(diagdds$Group, "5")
diagdds <- DESeq(
diagdds,
test = "Wald",
fitType = "parametric",
sfType = "poscounts" # <- important
)
resultsNames(diagdds)
res = results(diagdds, cooksCutoff = FALSE)
alpha = 0.05
sigtab = res[which(res$padj < alpha), ]
sigtab = cbind(as(sigtab, "data.frame"), as(phyloseq::tax_table(ps_deseq_sel)[rownames(sigtab), ], "matrix"))
# file base name
fname <- "DEGs_Group4_vs_Group5"
write.xlsx(sigtab, file = paste0(fname, ".xlsx"), rowNames = TRUE)
kable(sigtab) %>%
kable_styling(bootstrap_options = c("striped", "hover", "condensed", "responsive"))
library("ggplot2")
theme_set(theme_bw())
scale_fill_discrete <- function(palname = "Set1", ...) {
scale_fill_brewer(palette = palname, ...)
}
x = tapply(sigtab$log2FoldChange, sigtab$Order, function(x) max(x))
x = sort(x)
sigtab$Order = factor(as.character(sigtab$Order), levels=names(x))
x = tapply(sigtab$log2FoldChange, sigtab$Family, function(x) max(x))
x = sort(x)
sigtab$Family = factor(as.character(sigtab$Family), levels=names(x))
#ggplot(sigtab, aes(x=log2FoldChange, y=Family, color=Order)) + geom_point(aes(size=padj)) + scale_size_continuous(name="padj",range=c(8,4))+
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust=0.5))
# build the plot
p <- ggplot(sigtab, aes(x = log2FoldChange, y = Family, color = Order)) +
geom_point(aes(size = padj)) +
scale_size_continuous(name = "padj", range = c(8, 4)) +
theme_bw() +
theme(axis.text.x = element_text(angle = -25, hjust = 0, vjust = 0.5))
# SVG (svglite gives crisp text)
if (!requireNamespace("svglite", quietly = TRUE)) install.packages("svglite")
ggplot2::ggsave(paste0(fname, ".svg"), plot = p, device = svglite::svglite,
width = 8, height = 6, units = "in", dpi = 300)
# PNG
ggplot2::ggsave(paste0(fname, ".png"), plot = p, device = "png",
width = 8, height = 6, units = "in", dpi = 300)
knitr::include_graphics(paste0(fname, ".png"))
```