-
Vorgabe
#perform PCA analysis, Venn diagram analysis, as well as KEGG and GO annotations. We would also appreciate it if you could include CPM calculations for this dataset (gene_cpm_counts.xlsx). For comparative analysis, we are particularly interested in identifying DEGs between WT and ΔIJ across the different treatments and time points. I have already performed the six comparisons, using WT as the reference: ΔIJ-17 vs WT-17 – no treatment ΔIJ-24 vs WT-24 – no treatment preΔIJ-17 vs preWT-17 – Treatment A preΔIJ-24 vs preWT-24 – Treatment A 0_5ΔIJ-17 vs 0_5WT-17 – Treatment B 0_5ΔIJ-24 vs 0_5WT-24 – Treatment B To gain a deeper understanding of how the ∆adeIJ mutation influences response dynamics over time and under different stimuli, would you also be interested in the following additional comparisons? Within-strain treatment responses (to explore how each strain responds to treatments): WT: preWT-17 vs WT-17 → response to Treatment A at 17 h preWT-24 vs WT-24 → response to Treatment A at 24 h 0_5WT-17 vs WT-17 → response to Treatment B at 17 h 0_5WT-24 vs WT-24 → response to Treatment B at 24 h ∆adeIJ: preΔIJ-17 vs ΔIJ-17 → response to Treatment A at 17 h preΔIJ-24 vs ΔIJ-24 → response to Treatment A at 24 h 0_5ΔIJ-17 vs ΔIJ-17 → response to Treatment B at 17 h 0_5ΔIJ-24 vs ΔIJ-24 → response to Treatment B at 24 h Time-course comparisons (to investigate time-dependent changes within each condition): WT-24 vs WT-17 ΔIJ-24 vs ΔIJ-17 preWT-24 vs preWT-17 preΔIJ-24 vs preΔIJ-17 0_5WT-24 vs 0_5WT-17 0_5ΔIJ-24 vs 0_5ΔIJ-17 I reviewed the datasets again and noticed that there are no ∆adeAB samples included. Should we try to obtain ∆adeAB data from other datasets? However, I’m a bit concerned that batch effects might pose a challenge when integrating data from different datasets. > It is possible to analyze DEGs across various time points (17 and 24 h) and stimuli (treatment A and B, and without treatment) iswithin both the ∆adeIJ mutant and the WT strain as our phenotypic characterization of these strains across two times points and stimuli shows significant differences but the other mutant ∆adeAB (similar function as AdeIJ) shows no difference compared to WT, therefore we are wondering what's happened to ∆adeIJ. deltaIJ_17, WT_17 – ΔadeIJ and wildtype strains w/o exposure at 17 h (No treatment) deltaIJ_24, WT_24 – ΔadeIJ and wildtype strains w/o exposure at 24 h (No treatment) pre_deltaIJ_17, pre_WT_17 – ΔadeIJ and wildtype strains with 1 exposure at 17 h (Treatment A) pre_deltaIJ_24, pre_WT_24 – ΔadeIJ and wildtype strains with 1 exposure at 24 h (Treatment A) 0_5_deltaIJ_17, 0_5_WT_17 – ΔadeIJ and wildtype strains with 2 exposure at 17 h (Treatment B) 0_5_deltaIJ_24, 0_5_WT_24 – ΔadeIJ and wildtype strains with 2 exposure at 24 h (Treatment B) -
Preparing raw data
mkdir raw_data; cd raw_data ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-17-1/WT-17-1_1.fq.gz WT-17-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-17-1/WT-17-1_2.fq.gz WT-17-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-17-2/WT-17-2_1.fq.gz WT-17-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-17-2/WT-17-2_2.fq.gz WT-17-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-17-3/WT-17-3_1.fq.gz WT-17-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-17-3/WT-17-3_2.fq.gz WT-17-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-24-1/WT-24-1_1.fq.gz WT-24-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-24-1/WT-24-1_2.fq.gz WT-24-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-24-2/WT-24-2_1.fq.gz WT-24-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-24-2/WT-24-2_2.fq.gz WT-24-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-24-3/WT-24-3_1.fq.gz WT-24-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT-24-3/WT-24-3_2.fq.gz WT-24-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-17-1/ΔIJ-17-1_1.fq.gz deltaIJ-17-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-17-1/ΔIJ-17-1_2.fq.gz deltaIJ-17-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-17-2/ΔIJ-17-2_1.fq.gz deltaIJ-17-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-17-2/ΔIJ-17-2_2.fq.gz deltaIJ-17-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-17-3/ΔIJ-17-3_1.fq.gz deltaIJ-17-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-17-3/ΔIJ-17-3_2.fq.gz deltaIJ-17-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-24-1/ΔIJ-24-1_1.fq.gz deltaIJ-24-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-24-1/ΔIJ-24-1_2.fq.gz deltaIJ-24-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-24-2/ΔIJ-24-2_1.fq.gz deltaIJ-24-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-24-2/ΔIJ-24-2_2.fq.gz deltaIJ-24-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-24-3/ΔIJ-24-3_1.fq.gz deltaIJ-24-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/ΔIJ-24-3/ΔIJ-24-3_2.fq.gz deltaIJ-24-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-17-1/preWT-17-1_1.fq.gz pre_WT-17-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-17-1/preWT-17-1_2.fq.gz pre_WT-17-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-17-2/preWT-17-2_1.fq.gz pre_WT-17-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-17-2/preWT-17-2_2.fq.gz pre_WT-17-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-17-3/preWT-17-3_1.fq.gz pre_WT-17-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-17-3/preWT-17-3_2.fq.gz pre_WT-17-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-24-1/preWT-24-1_1.fq.gz pre_WT-24-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-24-1/preWT-24-1_2.fq.gz pre_WT-24-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-24-2/preWT-24-2_1.fq.gz pre_WT-24-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-24-2/preWT-24-2_2.fq.gz pre_WT-24-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-24-3/preWT-24-3_1.fq.gz pre_WT-24-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preWT-24-3/preWT-24-3_2.fq.gz pre_WT-24-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-17-1/preΔIJ-17-1_1.fq.gz pre_deltaIJ-17-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-17-1/preΔIJ-17-1_2.fq.gz pre_deltaIJ-17-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-17-2/preΔIJ-17-2_1.fq.gz pre_deltaIJ-17-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-17-2/preΔIJ-17-2_2.fq.gz pre_deltaIJ-17-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-17-3/preΔIJ-17-3_1.fq.gz pre_deltaIJ-17-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-17-3/preΔIJ-17-3_2.fq.gz pre_deltaIJ-17-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-24-1/preΔIJ-24-1_1.fq.gz pre_deltaIJ-24-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-24-1/preΔIJ-24-1_2.fq.gz pre_deltaIJ-24-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-24-2/preΔIJ-24-2_1.fq.gz pre_deltaIJ-24-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-24-2/preΔIJ-24-2_2.fq.gz pre_deltaIJ-24-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-24-3/preΔIJ-24-3_1.fq.gz pre_deltaIJ-24-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/preΔIJ-24-3/preΔIJ-24-3_2.fq.gz pre_deltaIJ-24-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-17-1/WT0_5-17-1_1.fq.gz 0_5_WT-17-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-17-1/WT0_5-17-1_2.fq.gz 0_5_WT-17-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-17-2/WT0_5-17-2_1.fq.gz 0_5_WT-17-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-17-2/WT0_5-17-2_2.fq.gz 0_5_WT-17-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-17-3/WT0_5-17-3_1.fq.gz 0_5_WT-17-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-17-3/WT0_5-17-3_2.fq.gz 0_5_WT-17-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-24-1/WT0_5-24-1_1.fq.gz 0_5_WT-24-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-24-1/WT0_5-24-1_2.fq.gz 0_5_WT-24-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-24-2/WT0_5-24-2_1.fq.gz 0_5_WT-24-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-24-2/WT0_5-24-2_2.fq.gz 0_5_WT-24-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-24-3/WT0_5-24-3_1.fq.gz 0_5_WT-24-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/WT0_5-24-3/WT0_5-24-3_2.fq.gz 0_5_WT-24-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-17-1/0_5ΔIJ-17-1_1.fq.gz 0_5_deltaIJ-17-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-17-1/0_5ΔIJ-17-1_2.fq.gz 0_5_deltaIJ-17-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-17-2/0_5ΔIJ-17-2_1.fq.gz 0_5_deltaIJ-17-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-17-2/0_5ΔIJ-17-2_2.fq.gz 0_5_deltaIJ-17-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-17-3/0_5ΔIJ-17-3_1.fq.gz 0_5_deltaIJ-17-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-17-3/0_5ΔIJ-17-3_2.fq.gz 0_5_deltaIJ-17-r3_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-24-1/0_5ΔIJ-24-1_1.fq.gz 0_5_deltaIJ-24-r1_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-24-1/0_5ΔIJ-24-1_2.fq.gz 0_5_deltaIJ-24-r1_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-24-2/0_5ΔIJ-24-2_1.fq.gz 0_5_deltaIJ-24-r2_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-24-2/0_5ΔIJ-24-2_2.fq.gz 0_5_deltaIJ-24-r2_R2.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-24-3/0_5ΔIJ-24-3_1.fq.gz 0_5_deltaIJ-24-r3_R1.fq.gz ln -s ../RSMR00204/X101SC25062155-Z01/X101SC25062155-Z01-J001/01.RawData/0_5ΔIJ-24-3/0_5ΔIJ-24-3_2.fq.gz 0_5_deltaIJ-24-r3_R2.fq.gz -
(Done) Downloading CP059040.fasta and CP059040.gff from GenBank
-
Preparing the directory trimmed
mkdir trimmed trimmed_unpaired; for sample_id in WT-17-r1 WT-17-r2 WT-17-r3 WT-24-r1 WT-24-r2 WT-24-r3 deltaIJ-17-r1 deltaIJ-17-r2 deltaIJ-17-r3 deltaIJ-24-r1 deltaIJ-24-r2 deltaIJ-24-r3 pre_WT-17-r1 pre_WT-17-r2 pre_WT-17-r3 pre_WT-24-r1 pre_WT-24-r2 pre_WT-24-r3 pre_deltaIJ-17-r1 pre_deltaIJ-17-r2 pre_deltaIJ-17-r3 pre_deltaIJ-24-r1 pre_deltaIJ-24-r2 pre_deltaIJ-24-r3 0_5_WT-17-r1 0_5_WT-17-r2 0_5_WT-17-r3 0_5_WT-24-r1 0_5_WT-24-r2 0_5_WT-24-r3 0_5_deltaIJ-17-r1 0_5_deltaIJ-17-r2 0_5_deltaIJ-17-r3 0_5_deltaIJ-24-r1 0_5_deltaIJ-24-r2 0_5_deltaIJ-24-r3; do \ java -jar /home/jhuang/Tools/Trimmomatic-0.36/trimmomatic-0.36.jar PE -threads 100 raw_data/${sample_id}_R1.fq.gz raw_data/${sample_id}_R2.fq.gz trimmed/${sample_id}_R1.fq.gz trimmed_unpaired/${sample_id}_R1.fq.gz trimmed/${sample_id}_R2.fq.gz trimmed_unpaired/${sample_id}_R2.fq.gz ILLUMINACLIP:/home/jhuang/Tools/Trimmomatic-0.36/adapters/TruSeq3-PE-2.fa:2:30:10:8:TRUE LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 AVGQUAL:20; done 2> trimmomatic_pe.log; done -
Preparing samplesheet.csv
sample,fastq_1,fastq_2,strandedness WT_17_r1,WT-17-r1_R1.fq.gz,WT-17-r1_R2.fq.gz,auto WT_17_r2,WT-17-r2_R1.fq.gz,WT-17-r2_R2.fq.gz,auto WT_17_r3,WT-17-r3_R1.fq.gz,WT-17-r3_R2.fq.gz,auto WT_24_r1,WT-24-r1_R1.fq.gz,WT-24-r1_R2.fq.gz,auto WT_24_r2,WT-24-r2_R1.fq.gz,WT-24-r2_R2.fq.gz,auto WT_24_r3,WT-24-r3_R1.fq.gz,WT-24-r3_R2.fq.gz,auto deltaIJ_17_r1,deltaIJ-17-r1_R1.fq.gz,deltaIJ-17-r1_R2.fq.gz,auto deltaIJ_17_r2,deltaIJ-17-r2_R1.fq.gz,deltaIJ-17-r2_R2.fq.gz,auto deltaIJ_17_r3,deltaIJ-17-r3_R1.fq.gz,deltaIJ-17-r3_R2.fq.gz,auto deltaIJ_24_r1,deltaIJ-24-r1_R1.fq.gz,deltaIJ-24-r1_R2.fq.gz,auto deltaIJ_24_r2,deltaIJ-24-r2_R1.fq.gz,deltaIJ-24-r2_R2.fq.gz,auto deltaIJ_24_r3,deltaIJ-24-r3_R1.fq.gz,deltaIJ-24-r3_R2.fq.gz,auto pre_WT_17_r1,pre_WT-17-r1_R1.fq.gz,pre_WT-17-r1_R2.fq.gz,auto pre_WT_17_r2,pre_WT-17-r2_R1.fq.gz,pre_WT-17-r2_R2.fq.gz,auto pre_WT_17_r3,pre_WT-17-r3_R1.fq.gz,pre_WT-17-r3_R2.fq.gz,auto pre_WT_24_r1,pre_WT-24-r1_R1.fq.gz,pre_WT-24-r1_R2.fq.gz,auto pre_WT_24_r2,pre_WT-24-r2_R1.fq.gz,pre_WT-24-r2_R2.fq.gz,auto pre_WT_24_r3,pre_WT-24-r3_R1.fq.gz,pre_WT-24-r3_R2.fq.gz,auto pre_deltaIJ_17_r1,pre_deltaIJ-17-r1_R1.fq.gz,pre_deltaIJ-17-r1_R2.fq.gz,auto pre_deltaIJ_17_r2,pre_deltaIJ-17-r2_R1.fq.gz,pre_deltaIJ-17-r2_R2.fq.gz,auto pre_deltaIJ_17_r3,pre_deltaIJ-17-r3_R1.fq.gz,pre_deltaIJ-17-r3_R2.fq.gz,auto pre_deltaIJ_24_r1,pre_deltaIJ-24-r1_R1.fq.gz,pre_deltaIJ-24-r1_R2.fq.gz,auto pre_deltaIJ_24_r2,pre_deltaIJ-24-r2_R1.fq.gz,pre_deltaIJ-24-r2_R2.fq.gz,auto pre_deltaIJ_24_r3,pre_deltaIJ-24-r3_R1.fq.gz,pre_deltaIJ-24-r3_R2.fq.gz,auto 0_5_WT_17_r1,0_5_WT-17-r1_R1.fq.gz,0_5_WT-17-r1_R2.fq.gz,auto 0_5_WT_17_r2,0_5_WT-17-r2_R1.fq.gz,0_5_WT-17-r2_R2.fq.gz,auto 0_5_WT_17_r3,0_5_WT-17-r3_R1.fq.gz,0_5_WT-17-r3_R2.fq.gz,auto 0_5_WT_24_r1,0_5_WT-24-r1_R1.fq.gz,0_5_WT-24-r1_R2.fq.gz,auto 0_5_WT_24_r2,0_5_WT-24-r2_R1.fq.gz,0_5_WT-24-r2_R2.fq.gz,auto 0_5_WT_24_r3,0_5_WT-24-r3_R1.fq.gz,0_5_WT-24-r3_R2.fq.gz,auto 0_5_deltaIJ_17_r1,0_5_deltaIJ-17-r1_R1.fq.gz,0_5_deltaIJ-17-r1_R2.fq.gz,auto 0_5_deltaIJ_17_r2,0_5_deltaIJ-17-r2_R1.fq.gz,0_5_deltaIJ-17-r2_R2.fq.gz,auto 0_5_deltaIJ_17_r3,0_5_deltaIJ-17-r3_R1.fq.gz,0_5_deltaIJ-17-r3_R2.fq.gz,auto 0_5_deltaIJ_24_r1,0_5_deltaIJ-24-r1_R1.fq.gz,0_5_deltaIJ-24-r1_R2.fq.gz,auto 0_5_deltaIJ_24_r2,0_5_deltaIJ-24-r2_R1.fq.gz,0_5_deltaIJ-24-r2_R2.fq.gz,auto 0_5_deltaIJ_24_r3,0_5_deltaIJ-24-r3_R1.fq.gz,0_5_deltaIJ-24-r3_R2.fq.gz,auto -
nextflow run
#Example1: http://xgenes.com/article/article-content/157/prepare-virus-gtf-for-nextflow-run/ docker pull nfcore/rnaseq ln -s /home/jhuang/Tools/nf-core-rnaseq-3.12.0/ rnaseq #Default: --gtf_group_features 'gene_id' --gtf_extra_attributes 'gene_name' --featurecounts_group_type 'gene_biotype' --featurecounts_feature_type 'exon' #(host_env) !NOT_WORKING! jhuang@WS-2290C:~/DATA/Data_Tam_RNAseq_2024$ /usr/local/bin/nextflow run rnaseq/main.nf --input samplesheet.csv --outdir results --fasta "/home/jhuang/DATA/Data_Tam_RNAseq_2024/CP059040.fasta" --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024/CP059040.gff" -profile docker -resume --max_cpus 55 --max_memory 512.GB --max_time 2400.h --save_align_intermeds --save_unaligned --save_reference --aligner 'star_salmon' --gtf_group_features 'gene_id' --gtf_extra_attributes 'gene_name' --featurecounts_group_type 'gene_biotype' --featurecounts_feature_type 'transcript' # -- DEBUG_1 (CDS --> exon in CP059040.gff) -- #Checking the record (see below) in results/genome/CP059040.gtf #In ./results/genome/CP059040.gtf e.g. "CP059040.1 Genbank transcript 1 1398 . + . transcript_id "gene-H0N29_00005"; gene_id "gene-H0N29_00005"; gene_name "dnaA"; Name "dnaA"; gbkey "Gene"; gene "dnaA"; gene_biotype "protein_coding"; locus_tag "H0N29_00005";" #--featurecounts_feature_type 'transcript' returns only the tRNA results #Since the tRNA records have "transcript and exon". In gene records, we have "transcript and CDS". replace the CDS with exon grep -P "\texon\t" CP059040.gff | sort | wc -l #96 grep -P "cmsearch\texon\t" CP059040.gff | wc -l #=10 ignal recognition particle sRNA small typ, transfer-messenger RNA, 5S ribosomal RNA grep -P "Genbank\texon\t" CP059040.gff | wc -l #=12 16S and 23S ribosomal RNA grep -P "tRNAscan-SE\texon\t" CP059040.gff | wc -l #tRNA 74 wc -l star_salmon/AUM_r3/quant.genes.sf #--featurecounts_feature_type 'transcript' results in 96 records! grep -P "\tCDS\t" CP059040.gff | wc -l #3701 sed 's/\tCDS\t/\texon\t/g' CP059040.gff > CP059040_m.gff grep -P "\texon\t" CP059040_m.gff | sort | wc -l #3797 # -- DEBUG_2: combination of 'CP059040_m.gff' and 'exon' results in ERROR, using 'transcript' instead! --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024/CP059040_m.gff" --featurecounts_feature_type 'transcript' # ---- SUCCESSFUL with directly downloaded gff3 and fasta from NCBI using docker after replacing 'CDS' with 'exon' ---- mv trimmed/*.fq.gz .; rmdir trimmed (host_env) /usr/local/bin/nextflow run rnaseq/main.nf --input samplesheet.csv --outdir results --fasta "/home/jhuang/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/CP059040.fasta" --gff "/home/jhuang/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/CP059040_m.gff" -profile docker -resume --max_cpus 90 --max_memory 900.GB --max_time 2400.h --save_align_intermeds --save_unaligned --save_reference --aligner 'star_salmon' --gtf_group_features 'gene_id' --gtf_extra_attributes 'gene_name' --featurecounts_group_type 'gene_biotype' --featurecounts_feature_type 'transcript' # -- DEBUG_3: make sure the header of fasta is the same to the *_m.gff file -
Prepare counts_fixed by hand: delete all “””, “gene-“, replace , to ‘\t’.
cp ./results/star_salmon/gene_raw_counts.csv counts.tsv #keep only gene_id cut -f1 -d',' counts.tsv > f1 cut -f3- -d',' counts.tsv > f3_ paste -d',' f1 f3_ > counts_fixed.tsv Rscript rna_timecourse_bacteria.R \ --counts counts_fixed.tsv \ --samples samples.tsv \ --condition_col condition \ --time_col time_h \ --emapper ~/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/eggnog_out.emapper.annotations.txt \ --volcano_csvs contrasts/ctrl_vs_treat.csv \ --outdir results_bacteria #Delete the repliate 2 of ΔadeIJ_two_17 and repliate 1 of ΔadeIJ_two_24 are outlier. paste -d$'\t' f1_32 f34 f36_ > counts_fixed_2.tsv Rscript rna_timecourse_bacteria.R \ --counts counts_fixed_2.tsv \ --samples samples_2.tsv \ --condition_col condition \ --time_col time_h \ --emapper ~/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/eggnog_out.emapper.annotations.txt \ --volcano_csvs contrasts/ctrl_vs_treat.csv \ --outdir results_bacteria_2 -
Import data and pca-plot
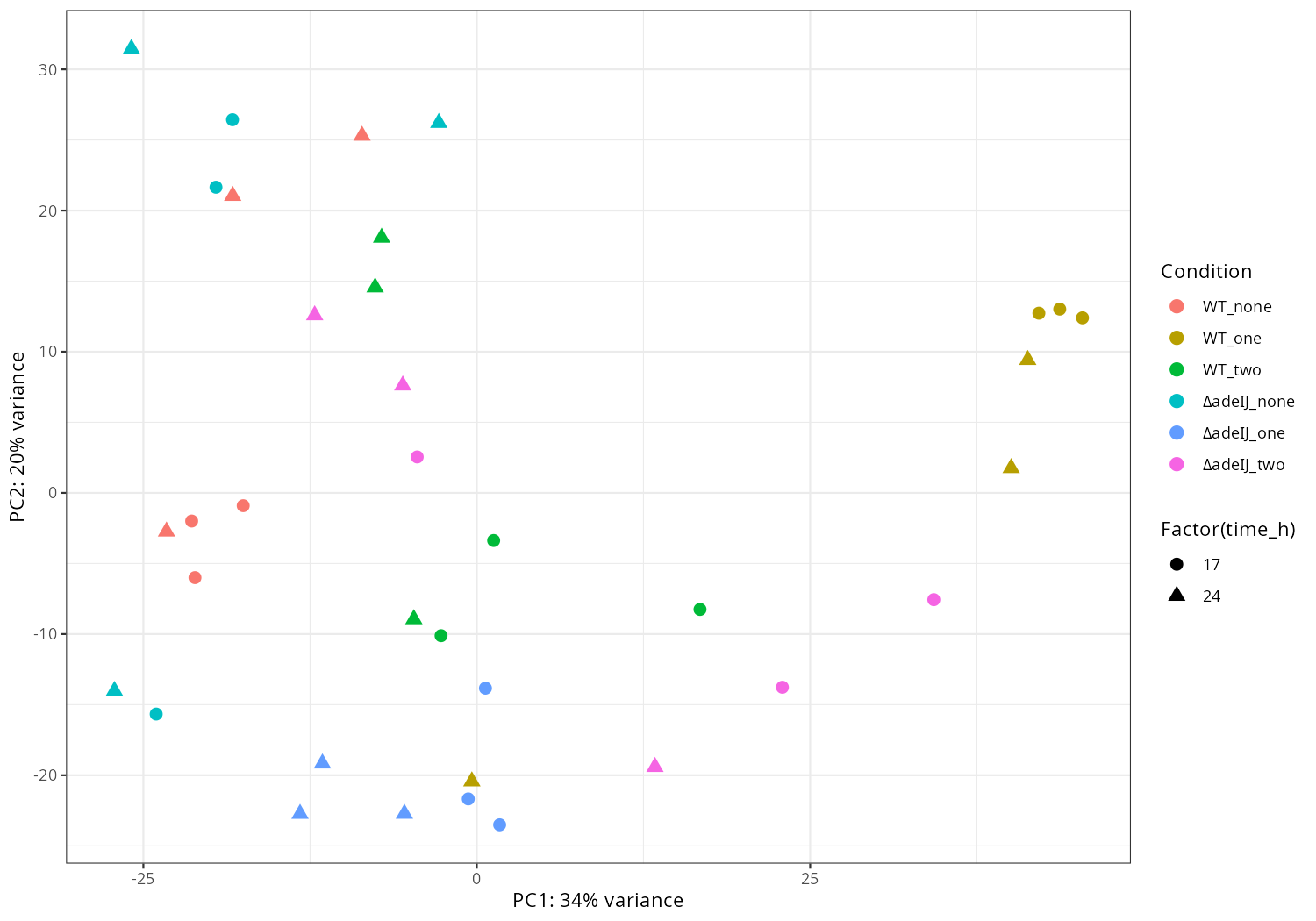
#mamba activate r_env #install.packages("ggfun") # Import the required libraries library("AnnotationDbi") library("clusterProfiler") library("ReactomePA") library(gplots) library(tximport) library(DESeq2) #library("org.Hs.eg.db") library(dplyr) library(tidyverse) #install.packages("devtools") #devtools::install_version("gtable", version = "0.3.0") library(gplots) library("RColorBrewer") #install.packages("ggrepel") library("ggrepel") # install.packages("openxlsx") library(openxlsx) library(EnhancedVolcano) library(DESeq2) library(edgeR) setwd("~/DATA/Data_Tam_RNAseq_2025_subMIC_exposure_ATCC19606/results/star_salmon") # Define paths to your Salmon output quantification files files <- c("WT_17_r1" = "./WT_17_r1/quant.sf", "WT_17_r2" = "./WT_17_r2/quant.sf", "WT_17_r3" = "./WT_17_r3/quant.sf", "WT_24_r1" = "./WT_24_r1/quant.sf", "WT_24_r2" = "./WT_24_r2/quant.sf", "WT_24_r3" = "./WT_24_r3/quant.sf", "deltaIJ_17_r1" = "./deltaIJ_17_r1/quant.sf", "deltaIJ_17_r2" = "./deltaIJ_17_r2/quant.sf", "deltaIJ_17_r3" = "./deltaIJ_17_r3/quant.sf", "deltaIJ_24_r1" = "./deltaIJ_24_r1/quant.sf", "deltaIJ_24_r2" = "./deltaIJ_24_r2/quant.sf", "deltaIJ_24_r3" = "./deltaIJ_24_r3/quant.sf", "pre_WT_17_r1" = "./pre_WT_17_r1/quant.sf", "pre_WT_17_r2" = "./pre_WT_17_r2/quant.sf", "pre_WT_17_r3" = "./pre_WT_17_r3/quant.sf", "pre_WT_24_r1" = "./pre_WT_24_r1/quant.sf", "pre_WT_24_r2" = "./pre_WT_24_r2/quant.sf", "pre_WT_24_r3" = "./pre_WT_24_r3/quant.sf", "pre_deltaIJ_17_r1" = "./pre_deltaIJ_17_r1/quant.sf", "pre_deltaIJ_17_r2" = "./pre_deltaIJ_17_r2/quant.sf", "pre_deltaIJ_17_r3" = "./pre_deltaIJ_17_r3/quant.sf", "pre_deltaIJ_24_r1" = "./pre_deltaIJ_24_r1/quant.sf", "pre_deltaIJ_24_r2" = "./pre_deltaIJ_24_r2/quant.sf", "pre_deltaIJ_24_r3" = "./pre_deltaIJ_24_r3/quant.sf", "0_5_WT_17_r1" = "./0_5_WT_17_r1/quant.sf", "0_5_WT_17_r2" = "./0_5_WT_17_r2/quant.sf", "0_5_WT_17_r3" = "./0_5_WT_17_r3/quant.sf", "0_5_WT_24_r1" = "./0_5_WT_24_r1/quant.sf", "0_5_WT_24_r2" = "./0_5_WT_24_r2/quant.sf", "0_5_WT_24_r3" = "./0_5_WT_24_r3/quant.sf", "0_5_deltaIJ_17_r1" = "./0_5_deltaIJ_17_r1/quant.sf", "0_5_deltaIJ_17_r2" = "./0_5_deltaIJ_17_r2/quant.sf", "0_5_deltaIJ_17_r3" = "./0_5_deltaIJ_17_r3/quant.sf", "0_5_deltaIJ_24_r1" = "./0_5_deltaIJ_24_r1/quant.sf", "0_5_deltaIJ_24_r2" = "./0_5_deltaIJ_24_r2/quant.sf", "0_5_deltaIJ_24_r3" = "./0_5_deltaIJ_24_r3/quant.sf") # Import the transcript abundance data with tximport txi <- tximport(files, type = "salmon", txIn = TRUE, txOut = TRUE) # Define the replicates and condition of the samples replicate <- factor(c("r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3", "r1", "r2", "r3")) condition <- factor(c("WT_none_17","WT_none_17","WT_none_17","WT_none_24","WT_none_24","WT_none_24", "deltaadeIJ_none_17","deltaadeIJ_none_17","deltaadeIJ_none_17","deltaadeIJ_none_24","deltaadeIJ_none_24","deltaadeIJ_none_24", "WT_one_17","WT_one_17","WT_one_17","WT_one_24","WT_one_24","WT_one_24", "deltaadeIJ_one_17","deltaadeIJ_one_17","deltaadeIJ_one_17","deltaadeIJ_one_24","deltaadeIJ_one_24","deltaadeIJ_one_24", "WT_two_17","WT_two_17","WT_two_17","WT_two_24","WT_two_24","WT_two_24", "deltaadeIJ_two_17","deltaadeIJ_two_17","deltaadeIJ_two_17","deltaadeIJ_two_24","deltaadeIJ_two_24","deltaadeIJ_two_24")) # Construct colData manually colData <- data.frame(condition=condition, replicate=replicate, row.names=names(files)) #dds <- DESeqDataSetFromTximport(txi, colData, design = ~ condition + batch) dds <- DESeqDataSetFromTximport(txi, colData, design = ~ condition) # -- Save the rlog-transformed counts -- dim(counts(dds)) head(counts(dds), 10) rld <- rlogTransformation(dds) rlog_counts <- assay(rld) write.xlsx(as.data.frame(rlog_counts), "gene_rlog_transformed_counts.xlsx") # -- pca -- png("pca2.png", 1200, 800) plotPCA(rld, intgroup=c("condition")) dev.off() png("pca3.png", 1200, 800) plotPCA(rld, intgroup=c("replicate")) dev.off() pdat <- plotPCA(rld, intgroup = c("condition","replicate"), returnData = TRUE) percentVar <- round(100 * attr(pdat, "percentVar")) # 1) keep only non-WT samples #pdat <- subset(pdat, !grepl("^WT_", condition)) # drop unused factor levels so empty WT facets disappear pdat$condition <- droplevels(pdat$condition) # 2) pretty condition names: deltaadeIJ -> ΔadeIJ pdat$condition <- gsub("^deltaadeIJ", "\u0394adeIJ", pdat$condition) png("pca4.png", 1200, 800) ggplot(pdat, aes(PC1, PC2, color = replicate)) + geom_point(size = 3) + facet_wrap(~ condition) + xlab(paste0("PC1: ", percentVar[1], "% variance")) + ylab(paste0("PC2: ", percentVar[2], "% variance")) + theme_classic() dev.off() pdat <- plotPCA(rld, intgroup = c("condition","replicate"), returnData = TRUE) percentVar <- round(100 * attr(pdat, "percentVar")) # Drop WT_* conditions from the data and from factor levels pdat <- subset(pdat, !grepl("^WT_", condition)) pdat$condition <- droplevels(pdat$condition) # Prettify condition labels for the legend: deltaadeIJ -> ΔadeIJ pdat$condition <- gsub("^deltaadeIJ", "\u0394adeIJ", pdat$condition) p <- ggplot(pdat, aes(PC1, PC2, color = replicate, shape = condition)) + geom_point(size = 3) + xlab(paste0("PC1: ", percentVar[1], "% variance")) + ylab(paste0("PC2: ", percentVar[2], "% variance")) + theme_classic() png("pca5.png", 1200, 800); print(p); dev.off() pdat <- plotPCA(rld, intgroup = c("condition","replicate"), returnData = TRUE) percentVar <- round(100 * attr(pdat, "percentVar")) p_fac <- ggplot(pdat, aes(PC1, PC2, color = replicate)) + geom_point(size = 3) + facet_wrap(~ condition) + xlab(paste0("PC1: ", percentVar[1], "% variance")) + ylab(paste0("PC2: ", percentVar[2], "% variance")) + theme_classic() png("pca6.png", 1200, 800); print(p_fac); dev.off() # -- heatmap -- png("heatmap2.png", 1200, 800) distsRL <- dist(t(assay(rld))) mat <- as.matrix(distsRL) hc <- hclust(distsRL) hmcol <- colorRampPalette(brewer.pal(9,"GnBu"))(100) heatmap.2(mat, Rowv=as.dendrogram(hc),symm=TRUE, trace="none",col = rev(hmcol), margin=c(13, 13)) dev.off() # -- pca_media_strain -- #png("pca_media.png", 1200, 800) #plotPCA(rld, intgroup=c("media")) #dev.off() #png("pca_strain.png", 1200, 800) #plotPCA(rld, intgroup=c("strain")) #dev.off() #png("pca_time.png", 1200, 800) #plotPCA(rld, intgroup=c("time")) #dev.off() -
Select the differentially expressed genes
#https://galaxyproject.eu/posts/2020/08/22/three-steps-to-galaxify-your-tool/ #https://www.biostars.org/p/282295/ #https://www.biostars.org/p/335751/ dds$condition [1] WT_none_17 WT_none_17 WT_none_17 WT_none_24 [5] WT_none_24 WT_none_24 deltaadeIJ_none_17 deltaadeIJ_none_17 [9] deltaadeIJ_none_17 deltaadeIJ_none_24 deltaadeIJ_none_24 deltaadeIJ_none_24 [13] WT_one_17 WT_one_17 WT_one_17 WT_one_24 [17] WT_one_24 WT_one_24 deltaadeIJ_one_17 deltaadeIJ_one_17 [21] deltaadeIJ_one_17 deltaadeIJ_one_24 deltaadeIJ_one_24 deltaadeIJ_one_24 [25] WT_two_17 WT_two_17 WT_two_17 WT_two_24 [29] WT_two_24 WT_two_24 deltaadeIJ_two_17 deltaadeIJ_two_17 [33] deltaadeIJ_two_17 deltaadeIJ_two_24 deltaadeIJ_two_24 deltaadeIJ_two_24 12 Levels: deltaadeIJ_none_17 deltaadeIJ_none_24 ... WT_two_24 #CONSOLE: mkdir star_salmon/degenes setwd("degenes") # Construct colData automatically sample_table <- data.frame( condition = condition, replicate = replicate ) split_cond <- do.call(rbind, strsplit(as.character(condition), "_")) colnames(split_cond) <- c("genotype", "exposure", "time") colData <- cbind(sample_table, split_cond) colData$genotype <- factor(colData$genotype) colData$exposure <- factor(colData$exposure) colData$time <- factor(colData$time) colData$group <- factor(paste(colData$genotype, colData$exposure, colData$time, sep = "_")) # Construct colData manually colData2 <- data.frame(condition=condition, row.names=names(files)) # 确保因子顺序(可选) colData$genotype <- relevel(factor(colData$genotype), ref = "WT") colData$exposure <- relevel(factor(colData$exposure), ref = "none") colData$time <- relevel(factor(colData$time), ref = "17") dds <- DESeqDataSetFromTximport(txi, colData, design = ~ genotype * exposure * time) dds <- DESeq(dds, betaPrior = FALSE) resultsNames(dds) [1] "Intercept" [2] "genotype_deltaadeIJ_vs_WT" [3] "exposure_one_vs_none" [4] "exposure_two_vs_none" [5] "time_24_vs_17" [6] "genotypedeltaadeIJ.exposureone" [7] "genotypedeltaadeIJ.exposuretwo" [8] "genotypedeltaadeIJ.time24" [9] "exposureone.time24" [10] "exposuretwo.time24" [11] "genotypedeltaadeIJ.exposureone.time24" [12] "genotypedeltaadeIJ.exposuretwo.time24" # 提取 genotype 的主效应: up 10, down 4 contrast <- "genotype_deltaadeIJ_vs_WT" res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(contrast, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(contrast, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(contrast, "down.txt", sep="-")) # 提取 one exposure 的主效应: up 196; down 298 contrast <- "exposure_one_vs_none" res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(contrast, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(contrast, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(contrast, "down.txt", sep="-")) # 提取 two exposure 的主效应: up 80; down 105 contrast <- "exposure_two_vs_none" res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(contrast, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(contrast, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(contrast, "down.txt", sep="-")) # 提取 time 的主效应 up 10; down 2 contrast <- "time_24_vs_17" res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(contrast, "all.txt", sep="-")) up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(contrast, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(contrast, "down.txt", sep="-")) #1.) ΔadeIJ_none 17h vs WT_none 17h #2.) ΔadeIJ_none 24h vs WT_none 24h #3.) ΔadeIJ_one 17h vs WT_one 17h #4.) ΔadeIJ_one 24h vs WT_one 24h #5.) ΔadeIJ_two 17h vs WT_two 17h #6.) ΔadeIJ_two 24h vs WT_two 24h #---- relevel to control ---- dds <- DESeqDataSetFromTximport(txi, colData, design = ~ condition) dds$condition <- relevel(dds$condition, "WT_none_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_none_17_vs_WT_none_17") dds$condition <- relevel(dds$condition, "WT_none_24") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_none_24_vs_WT_none_24") dds$condition <- relevel(dds$condition, "WT_one_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_one_17_vs_WT_one_17") dds$condition <- relevel(dds$condition, "WT_one_24") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_one_24_vs_WT_one_24") dds$condition <- relevel(dds$condition, "WT_two_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_two_17_vs_WT_two_17") dds$condition <- relevel(dds$condition, "WT_two_24") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_two_24_vs_WT_two_24") # WT_none_xh dds$condition <- relevel(dds$condition, "WT_none_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("WT_none_24_vs_WT_none_17") # WT_one_xh dds$condition <- relevel(dds$condition, "WT_one_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("WT_one_24_vs_WT_one_17") # WT_two_xh dds$condition <- relevel(dds$condition, "WT_two_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("WT_two_24_vs_WT_two_17") # deltaadeIJ_none_xh dds$condition <- relevel(dds$condition, "deltaadeIJ_none_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_none_24_vs_deltaadeIJ_none_17") # deltaadeIJ_one_xh dds$condition <- relevel(dds$condition, "deltaadeIJ_one_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_one_24_vs_deltaadeIJ_one_17") # deltaadeIJ_two_xh dds$condition <- relevel(dds$condition, "deltaadeIJ_two_17") dds = DESeq(dds, betaPrior=FALSE) resultsNames(dds) clist <- c("deltaadeIJ_two_24_vs_deltaadeIJ_two_17") for (i in clist) { contrast = paste("condition", i, sep="_") #for_Mac_vs_LB contrast = paste("media", i, sep="_") res = results(dds, name=contrast) res <- res[!is.na(res$log2FoldChange),] res_df <- as.data.frame(res) write.csv(as.data.frame(res_df[order(res_df$pvalue),]), file = paste(i, "all.txt", sep="-")) #res$log2FoldChange < -2 & res$padj < 5e-2 up <- subset(res_df, padj<=0.05 & log2FoldChange>=2) down <- subset(res_df, padj<=0.05 & log2FoldChange<=-2) write.csv(as.data.frame(up[order(up$log2FoldChange,decreasing=TRUE),]), file = paste(i, "up.txt", sep="-")) write.csv(as.data.frame(down[order(abs(down$log2FoldChange),decreasing=TRUE),]), file = paste(i, "down.txt", sep="-")) } # -- Under host-env (mamba activate plot-numpy1) -- mamba activate plot-numpy1 grep -P "\tgene\t" CP059040_m.gff > CP059040_gene.gff for cmp in deltaadeIJ_none_17_vs_WT_none_17 deltaadeIJ_none_24_vs_WT_none_24 deltaadeIJ_one_17_vs_WT_one_17 deltaadeIJ_one_24_vs_WT_one_24 deltaadeIJ_two_17_vs_WT_two_17 deltaadeIJ_two_24_vs_WT_two_24 WT_none_24_vs_WT_none_17 WT_one_24_vs_WT_one_17 WT_two_24_vs_WT_two_17 deltaadeIJ_none_24_vs_deltaadeIJ_none_17 deltaadeIJ_one_24_vs_deltaadeIJ_one_17 deltaadeIJ_two_24_vs_deltaadeIJ_two_17; do python3 ~/Scripts/replace_gene_names.py /home/jhuang/DATA/Data_Tam_RNAseq_2025_subMIC_exposure_ATCC19606/CP059040_gene.gff ${cmp}-all.txt ${cmp}-all.csv python3 ~/Scripts/replace_gene_names.py /home/jhuang/DATA/Data_Tam_RNAseq_2025_subMIC_exposure_ATCC19606/CP059040_gene.gff ${cmp}-up.txt ${cmp}-up.csv python3 ~/Scripts/replace_gene_names.py /home/jhuang/DATA/Data_Tam_RNAseq_2025_subMIC_exposure_ATCC19606/CP059040_gene.gff ${cmp}-down.txt ${cmp}-down.csv done #deltaadeIJ_none_24_vs_deltaadeIJ_none_17 up(0) down(0) #deltaadeIJ_one_24_vs_deltaadeIJ_one_17 up(0) down(8: gabT, H0N29_11475, H0N29_01015, H0N29_01030, ...) #deltaadeIJ_two_24_vs_deltaadeIJ_two_17 up(8) down(51) -
(NOT_PERFORMED) Volcano plots
# ---- delta sbp TSB 2h vs WT TSB 2h ---- res <- read.csv("deltasbp_TSB_2h_vs_WT_TSB_2h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] #print(duplicated_genes) # [1] "bfr" "lipA" "ahpF" "pcaF" "alr" "pcaD" "cydB" "lpdA" "pgaC" "ppk1" #[11] "pcaF" "tuf" "galE" "murI" "yccS" "rrf" "rrf" "arsB" "ptsP" "umuD" #[21] "map" "pgaB" "rrf" "rrf" "rrf" "pgaD" "uraH" "benE" #res[res$GeneName == "bfr", ] #1st_strategy First occurrence is kept and Subsequent duplicates are removed #res <- res[!duplicated(res$GeneName), ] #2nd_strategy keep the row with the smallest padj value for each GeneName res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Assuming res is your dataframe and already processed # Filter up-regulated genes: log2FoldChange > 2 and padj < 5e-2 up_regulated <- res[res$log2FoldChange > 2 & res$padj < 5e-2, ] # Filter down-regulated genes: log2FoldChange < -2 and padj < 5e-2 down_regulated <- res[res$log2FoldChange < -2 & res$padj < 5e-2, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet addWorksheet(wb, "Complete_Data") writeData(wb, "Complete_Data", res) # Add the up-regulated genes as the second sheet addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file saveWorkbook(wb, "Gene_Expression_Δsbp_TSB_2h_vs_WT_TSB_2h.xlsx", overwrite = TRUE) # Set the 'GeneName' column as row.names rownames(res) <- res$GeneName # Drop the 'GeneName' column since it's now the row names res$GeneName <- NULL head(res) ## Ensure the data frame matches the expected format ## For example, it should have columns: log2FoldChange, padj, etc. #res <- as.data.frame(res) ## Remove rows with NA in log2FoldChange (if needed) #res <- res[!is.na(res$log2FoldChange),] # Replace padj = 0 with a small value #NO_SUCH_RECORDS: res$padj[res$padj == 0] <- 1e-150 #library(EnhancedVolcano) # Assuming res is already sorted and processed png("Δsbp_TSB_2h_vs_WT_TSB_2h.png", width=1200, height=1200) #max.overlaps = 10 EnhancedVolcano(res, lab = rownames(res), x = 'log2FoldChange', y = 'padj', pCutoff = 5e-2, FCcutoff = 2, title = '', subtitleLabSize = 18, pointSize = 3.0, labSize = 5.0, colAlpha = 1, legendIconSize = 4.0, drawConnectors = TRUE, widthConnectors = 0.5, colConnectors = 'black', subtitle = expression("Δsbp TSB 2h versus WT TSB 2h")) dev.off() # ---- delta sbp TSB 4h vs WT TSB 4h ---- res <- read.csv("deltasbp_TSB_4h_vs_WT_TSB_4h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Assuming res is your dataframe and already processed # Filter up-regulated genes: log2FoldChange > 2 and padj < 5e-2 up_regulated <- res[res$log2FoldChange > 2 & res$padj < 5e-2, ] # Filter down-regulated genes: log2FoldChange < -2 and padj < 5e-2 down_regulated <- res[res$log2FoldChange < -2 & res$padj < 5e-2, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet addWorksheet(wb, "Complete_Data") writeData(wb, "Complete_Data", res) # Add the up-regulated genes as the second sheet addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file saveWorkbook(wb, "Gene_Expression_Δsbp_TSB_4h_vs_WT_TSB_4h.xlsx", overwrite = TRUE) # Set the 'GeneName' column as row.names rownames(res) <- res$GeneName # Drop the 'GeneName' column since it's now the row names res$GeneName <- NULL head(res) #library(EnhancedVolcano) # Assuming res is already sorted and processed png("Δsbp_TSB_4h_vs_WT_TSB_4h.png", width=1200, height=1200) #max.overlaps = 10 EnhancedVolcano(res, lab = rownames(res), x = 'log2FoldChange', y = 'padj', pCutoff = 5e-2, FCcutoff = 2, title = '', subtitleLabSize = 18, pointSize = 3.0, labSize = 5.0, colAlpha = 1, legendIconSize = 4.0, drawConnectors = TRUE, widthConnectors = 0.5, colConnectors = 'black', subtitle = expression("Δsbp TSB 4h versus WT TSB 4h")) dev.off() # ---- delta sbp TSB 18h vs WT TSB 18h ---- res <- read.csv("deltasbp_TSB_18h_vs_WT_TSB_18h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Assuming res is your dataframe and already processed # Filter up-regulated genes: log2FoldChange > 2 and padj < 5e-2 up_regulated <- res[res$log2FoldChange > 2 & res$padj < 5e-2, ] # Filter down-regulated genes: log2FoldChange < -2 and padj < 5e-2 down_regulated <- res[res$log2FoldChange < -2 & res$padj < 5e-2, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet addWorksheet(wb, "Complete_Data") writeData(wb, "Complete_Data", res) # Add the up-regulated genes as the second sheet addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file saveWorkbook(wb, "Gene_Expression_Δsbp_TSB_18h_vs_WT_TSB_18h.xlsx", overwrite = TRUE) # Set the 'GeneName' column as row.names rownames(res) <- res$GeneName # Drop the 'GeneName' column since it's now the row names res$GeneName <- NULL head(res) #library(EnhancedVolcano) # Assuming res is already sorted and processed png("Δsbp_TSB_18h_vs_WT_TSB_18h.png", width=1200, height=1200) #max.overlaps = 10 EnhancedVolcano(res, lab = rownames(res), x = 'log2FoldChange', y = 'padj', pCutoff = 5e-2, FCcutoff = 2, title = '', subtitleLabSize = 18, pointSize = 3.0, labSize = 5.0, colAlpha = 1, legendIconSize = 4.0, drawConnectors = TRUE, widthConnectors = 0.5, colConnectors = 'black', subtitle = expression("Δsbp TSB 18h versus WT TSB 18h")) dev.off() # ---- delta sbp MH 2h vs WT MH 2h ---- res <- read.csv("deltasbp_MH_2h_vs_WT_MH_2h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] #print(duplicated_genes) # [1] "bfr" "lipA" "ahpF" "pcaF" "alr" "pcaD" "cydB" "lpdA" "pgaC" "ppk1" #[11] "pcaF" "tuf" "galE" "murI" "yccS" "rrf" "rrf" "arsB" "ptsP" "umuD" #[21] "map" "pgaB" "rrf" "rrf" "rrf" "pgaD" "uraH" "benE" #res[res$GeneName == "bfr", ] #1st_strategy First occurrence is kept and Subsequent duplicates are removed #res <- res[!duplicated(res$GeneName), ] #2nd_strategy keep the row with the smallest padj value for each GeneName res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Assuming res is your dataframe and already processed # Filter up-regulated genes: log2FoldChange > 2 and padj < 5e-2 up_regulated <- res[res$log2FoldChange > 2 & res$padj < 5e-2, ] # Filter down-regulated genes: log2FoldChange < -2 and padj < 5e-2 down_regulated <- res[res$log2FoldChange < -2 & res$padj < 5e-2, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet addWorksheet(wb, "Complete_Data") writeData(wb, "Complete_Data", res) # Add the up-regulated genes as the second sheet addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file saveWorkbook(wb, "Gene_Expression_Δsbp_MH_2h_vs_WT_MH_2h.xlsx", overwrite = TRUE) # Set the 'GeneName' column as row.names rownames(res) <- res$GeneName # Drop the 'GeneName' column since it's now the row names res$GeneName <- NULL head(res) ## Ensure the data frame matches the expected format ## For example, it should have columns: log2FoldChange, padj, etc. #res <- as.data.frame(res) ## Remove rows with NA in log2FoldChange (if needed) #res <- res[!is.na(res$log2FoldChange),] # Replace padj = 0 with a small value #NO_SUCH_RECORDS: res$padj[res$padj == 0] <- 1e-150 #library(EnhancedVolcano) # Assuming res is already sorted and processed png("Δsbp_MH_2h_vs_WT_MH_2h.png", width=1200, height=1200) #max.overlaps = 10 EnhancedVolcano(res, lab = rownames(res), x = 'log2FoldChange', y = 'padj', pCutoff = 5e-2, FCcutoff = 2, title = '', subtitleLabSize = 18, pointSize = 3.0, labSize = 5.0, colAlpha = 1, legendIconSize = 4.0, drawConnectors = TRUE, widthConnectors = 0.5, colConnectors = 'black', subtitle = expression("Δsbp MH 2h versus WT MH 2h")) dev.off() # ---- delta sbp MH 4h vs WT MH 4h ---- res <- read.csv("deltasbp_MH_4h_vs_WT_MH_4h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Assuming res is your dataframe and already processed # Filter up-regulated genes: log2FoldChange > 2 and padj < 5e-2 up_regulated <- res[res$log2FoldChange > 2 & res$padj < 5e-2, ] # Filter down-regulated genes: log2FoldChange < -2 and padj < 5e-2 down_regulated <- res[res$log2FoldChange < -2 & res$padj < 5e-2, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet addWorksheet(wb, "Complete_Data") writeData(wb, "Complete_Data", res) # Add the up-regulated genes as the second sheet addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file saveWorkbook(wb, "Gene_Expression_Δsbp_MH_4h_vs_WT_MH_4h.xlsx", overwrite = TRUE) # Set the 'GeneName' column as row.names rownames(res) <- res$GeneName # Drop the 'GeneName' column since it's now the row names res$GeneName <- NULL head(res) #library(EnhancedVolcano) # Assuming res is already sorted and processed png("Δsbp_MH_4h_vs_WT_MH_4h.png", width=1200, height=1200) #max.overlaps = 10 EnhancedVolcano(res, lab = rownames(res), x = 'log2FoldChange', y = 'padj', pCutoff = 5e-2, FCcutoff = 2, title = '', subtitleLabSize = 18, pointSize = 3.0, labSize = 5.0, colAlpha = 1, legendIconSize = 4.0, drawConnectors = TRUE, widthConnectors = 0.5, colConnectors = 'black', subtitle = expression("Δsbp MH 4h versus WT MH 4h")) dev.off() # ---- delta sbp MH 18h vs WT MH 18h ---- res <- read.csv("deltasbp_MH_18h_vs_WT_MH_18h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Assuming res is your dataframe and already processed # Filter up-regulated genes: log2FoldChange > 2 and padj < 5e-2 up_regulated <- res[res$log2FoldChange > 2 & res$padj < 5e-2, ] # Filter down-regulated genes: log2FoldChange < -2 and padj < 5e-2 down_regulated <- res[res$log2FoldChange < -2 & res$padj < 5e-2, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet addWorksheet(wb, "Complete_Data") writeData(wb, "Complete_Data", res) # Add the up-regulated genes as the second sheet addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file saveWorkbook(wb, "Gene_Expression_Δsbp_MH_18h_vs_WT_MH_18h.xlsx", overwrite = TRUE) # Set the 'GeneName' column as row.names rownames(res) <- res$GeneName # Drop the 'GeneName' column since it's now the row names res$GeneName <- NULL head(res) #library(EnhancedVolcano) # Assuming res is already sorted and processed png("Δsbp_MH_18h_vs_WT_MH_18h.png", width=1200, height=1200) #max.overlaps = 10 EnhancedVolcano(res, lab = rownames(res), x = 'log2FoldChange', y = 'padj', pCutoff = 5e-2, FCcutoff = 2, title = '', subtitleLabSize = 18, pointSize = 3.0, labSize = 5.0, colAlpha = 1, legendIconSize = 4.0, drawConnectors = TRUE, widthConnectors = 0.5, colConnectors = 'black', subtitle = expression("Δsbp MH 18h versus WT MH 18h")) dev.off() #Annotate the Gene_Expression_xxx_vs_yyy.xlsx in the next steps (see below e.g. Gene_Expression_with_Annotations_Urine_vs_MHB.xlsx) -
Clustering the genes and draw heatmap
#http://xgenes.com/article/article-content/150/draw-venn-diagrams-using-matplotlib/ #http://xgenes.com/article/article-content/276/go-terms-for-s-epidermidis/ # save the Up-regulated and Down-regulated genes into -up.id and -down.id for i in deltaadeIJ_none_17_vs_WT_none_17 deltaadeIJ_none_24_vs_WT_none_24 deltaadeIJ_one_17_vs_WT_one_17 deltaadeIJ_one_24_vs_WT_one_24 deltaadeIJ_two_17_vs_WT_two_17 deltaadeIJ_two_24_vs_WT_two_24 WT_none_24_vs_WT_none_17 WT_one_24_vs_WT_one_17 WT_two_24_vs_WT_two_17 deltaadeIJ_none_24_vs_deltaadeIJ_none_17 deltaadeIJ_one_24_vs_deltaadeIJ_one_17 deltaadeIJ_two_24_vs_deltaadeIJ_two_17; do echo "cut -d',' -f1-1 ${i}-up.txt > ${i}-up.id"; echo "cut -d',' -f1-1 ${i}-down.txt > ${i}-down.id"; done # ------------------ Heatmap generation for two samples ---------------------- ## ------------------------------------------------------------ ## DEGs heatmap (dynamic GOI + dynamic column tags) ## Example contrast: deltasbp_TSB_2h_vs_WT_TSB_2h ## Assumes 'rld' (or 'vsd') is in the environment (DESeq2 transform) ## ------------------------------------------------------------ #12 deltaIJ_17_vs_WT_17-up.txt #1 deltaIJ_24_vs_WT_24-up.txt #239 pre_deltaIJ_17_vs_pre_WT_17-up.txt #84 pre_deltaIJ_24_vs_pre_WT_24-up.txt #75 0_5_deltaIJ_17_vs_0_5_WT_17-up.txt #2 0_5_deltaIJ_24_vs_0_5_WT_24-up.txt #4 deltaIJ_17_vs_WT_17-down.txt #3 deltaIJ_24_vs_WT_24-down.txt #91 pre_deltaIJ_17_vs_pre_WT_17-down.txt #65 pre_deltaIJ_24_vs_pre_WT_24-down.txt #15 0_5_deltaIJ_17_vs_0_5_WT_17-down.txt #4 0_5_deltaIJ_24_vs_0_5_WT_24-down.txt ## 0) Config --------------------------------------------------- contrast <- "deltaadeIJ_none_17_vs_WT_none_17" #up 11, down 3 vs. (10,4) contrast <- "deltaadeIJ_none_24_vs_WT_none_24" #up 0, down 2 vs. (0,2) contrast <- "deltaadeIJ_one_17_vs_WT_one_17" #up 238, down 90 vs. (239,89) --> height 2600 contrast <- "deltaadeIJ_one_24_vs_WT_one_24" #up 83, down 64 vs. (64,71) --> height 1600 contrast <- "deltaadeIJ_two_17_vs_WT_two_17" #up 74, down 14 vs. (75,9) --> height 1000 contrast <- "deltaadeIJ_two_24_vs_WT_two_24" #up 1, down 3 vs. (3,3) contrast <- "WT_none_24_vs_WT_none_17" #(up 10, down 2) contrast <- "WT_one_24_vs_WT_one_17" #(up 97, down 3) contrast <- "WT_two_24_vs_WT_two_17" #(up 12, down 1) contrast <- "deltaadeIJ_none_24_vs_deltaadeIJ_none_17" #(up 0, down 0) contrast <- "deltaadeIJ_one_24_vs_deltaadeIJ_one_17" #(up 0, down 10) contrast <- "deltaadeIJ_two_24_vs_deltaadeIJ_two_17" #(up 8, down 51) ## 1) Packages ------------------------------------------------- need <- c("gplots") to_install <- setdiff(need, rownames(installed.packages())) if (length(to_install)) install.packages(to_install, repos = "https://cloud.r-project.org") suppressPackageStartupMessages(library(gplots)) ## 2) Helpers -------------------------------------------------- # Read IDs from a file that may be: # - one column with or without header "Gene_Id" # - may contain quotes read_ids_from_file <- function(path) { if (!file.exists(path)) stop("File not found: ", path) df <- tryCatch( read.table(path, header = TRUE, stringsAsFactors = FALSE, quote = "\"'", comment.char = ""), error = function(e) NULL ) if (!is.null(df) && ncol(df) >= 1) { if ("Gene_Id" %in% names(df)) { ids <- df[["Gene_Id"]] } else if (ncol(df) == 1L) { ids <- df[[1]] } else { first_nonempty <- which(colSums(df != "", na.rm = TRUE) > 0)[1] if (is.na(first_nonempty)) stop("No usable IDs in: ", path) ids <- df[[first_nonempty]] } } else { df2 <- read.table(path, header = FALSE, stringsAsFactors = FALSE, quote = "\"'", comment.char = "") if (ncol(df2) < 1L) stop("No usable IDs in: ", path) ids <- df2[[1]] } ids <- trimws(gsub('"', "", ids)) ids[nzchar(ids)] } #BREAK_LINE # From "A_vs_B" get c("A","B") split_contrast_groups <- function(x) { parts <- strsplit(x, "_vs_", fixed = TRUE)[[1]] if (length(parts) != 2L) stop("Contrast must be in the form 'GroupA_vs_GroupB'") parts } # Match whole tags at boundaries or underscores match_tags <- function(nms, tags) { pat <- paste0("(^|_)(?:", paste(tags, collapse = "|"), ")(_|$)") grepl(pat, nms, perl = TRUE) } ## 3) Expression matrix (DESeq2 rlog/vst) ---------------------- # Use rld if present; otherwise try vsd if (exists("rld")) { expr_all <- assay(rld) } else if (exists("vsd")) { expr_all <- assay(vsd) } else { stop("Neither 'rld' nor 'vsd' object is available in the environment.") } RNASeq.NoCellLine <- as.matrix(expr_all) colnames(RNASeq.NoCellLine) <- c("WT_none_17_r1", "WT_none_17_r2", "WT_none_17_r3", "WT_none_24_r1", "WT_none_24_r2", "WT_none_24_r3", "deltaadeIJ_none_17_r1", "deltaadeIJ_none_17_r2", "deltaadeIJ_none_17_r3", "deltaadeIJ_none_24_r1", "deltaadeIJ_none_24_r2", "deltaadeIJ_none_24_r3", "WT_one_17_r1", "WT_one_17_r2", "WT_one_17_r3", "WT_one_24_r1", "WT_one_24_r2", "WT_one_24_r3", "deltaadeIJ_one_17_r1", "deltaadeIJ_one_17_r2", "deltaadeIJ_one_17_r3", "deltaadeIJ_one_24_r1", "deltaadeIJ_one_24_r2", "deltaadeIJ_one_24_r3", "WT_two_17_r1", "WT_two_17_r2", "WT_two_17_r3", "WT_two_24_r1", "WT_two_24_r2", "WT_two_24_r3", "deltaadeIJ_two_17_r1", "deltaadeIJ_two_17_r2", "deltaadeIJ_two_17_r3", "deltaadeIJ_two_24_r1", "deltaadeIJ_two_24_r2", "deltaadeIJ_two_24_r3") # -- RUN the code with the new contract from HERE after first run -- ## 4) Build GOI from the two .id files (Note that if empty not run!)------------------------- up_file <- paste0(contrast, "-up.id") down_file <- paste0(contrast, "-down.id") GOI_up <- read_ids_from_file(up_file) GOI_down <- read_ids_from_file(down_file) GOI <- unique(c(GOI_up, GOI_down)) if (length(GOI) == 0) stop("No gene IDs found in up/down .id files.") # GOI are already 'gene-*' in your data — use them directly for matching present <- intersect(rownames(RNASeq.NoCellLine), GOI) if (!length(present)) stop("None of the GOI were found among the rownames of the expression matrix.") # Optional: report truly missing IDs (on the same 'gene-*' format) missing <- setdiff(GOI, present) if (length(missing)) message("Note: ", length(missing), " GOI not found and will be skipped.") ## 5) Keep ONLY columns for the two groups in the contrast ----- groups <- split_contrast_groups(contrast) # e.g., c("deltasbp_TSB_2h", "WT_TSB_2h") keep_cols <- match_tags(colnames(RNASeq.NoCellLine), groups) if (!any(keep_cols)) { stop("No columns matched the contrast groups: ", paste(groups, collapse = " and "), ". Check your column names or implement colData-based filtering.") } cols_idx <- which(keep_cols) sub_colnames <- colnames(RNASeq.NoCellLine)[cols_idx] # Put the second group first (e.g., WT first in 'deltasbp..._vs_WT...') ord <- order(!grepl(paste0("(^|_)", groups[2], "(_|$)"), sub_colnames, perl = TRUE)) # Subset safely expr_sub <- RNASeq.NoCellLine[present, cols_idx, drop = FALSE][, ord, drop = FALSE] ## 6) Remove constant/NA rows ---------------------------------- row_ok <- apply(expr_sub, 1, function(x) is.finite(sum(x)) && var(x, na.rm = TRUE) > 0) if (any(!row_ok)) message("Removing ", sum(!row_ok), " constant/NA rows.") datamat <- expr_sub[row_ok, , drop = FALSE] # Save the filtered matrix used for the heatmap (optional) out_mat <- paste0("DEGs_heatmap_expression_data_", contrast, ".txt") write.csv(as.data.frame(datamat), file = out_mat, quote = FALSE) ## 6.1) Pretty labels (display only) --------------------------- # Row labels: strip 'gene-' labRow_pretty <- sub("^gene-", "", rownames(datamat)) # Column labels: 'deltaadeIJ' -> 'ΔadeIJ' and nicer spacing labCol_pretty <- colnames(datamat) labCol_pretty <- gsub("^deltaadeIJ", "\u0394adeIJ", labCol_pretty) labCol_pretty <- gsub("_", " ", labCol_pretty) # e.g., WT_TSB_2h_r1 -> "WT TSB 2h r1" # If you prefer to drop replicate suffixes, uncomment: # labCol_pretty <- gsub(" r\\d+$", "", labCol_pretty) ## 7) Clustering ----------------------------------------------- # Row clustering with Pearson distance hr <- hclust(as.dist(1-cor(t(datamat), method="pearson")), method="complete") #row_cor <- suppressWarnings(cor(t(datamat), method = "pearson", use = "pairwise.complete.obs")) #row_cor[!is.finite(row_cor)] <- 0 #hr <- hclust(as.dist(1 - row_cor), method = "complete") # Color row-side groups by cutting the dendrogram mycl <- cutree(hr, h = max(hr$height) / 1.1) palette_base <- c("yellow","blue","orange","magenta","cyan","red","green","maroon", "lightblue","pink","purple","lightcyan","salmon","lightgreen") mycol <- palette_base[(as.vector(mycl) - 1) %% length(palette_base) + 1] #BREAK_LINE png(paste0("DEGs_heatmap_", contrast, ".png"), width=800, height=600) heatmap.2(datamat, Rowv = as.dendrogram(hr), col = bluered(75), scale = "row", RowSideColors = mycol, trace = "none", margin = c(10, 20), # bottom, left sepwidth = c(0, 0), dendrogram = 'row', Colv = 'false', density.info = 'none', labRow = labRow_pretty, # row labels WITHOUT "gene-" labCol = labCol_pretty, # col labels with Δsbp + spaces cexRow = 2.5, cexCol = 2.5, srtCol = 15, lhei = c(0.6, 4), # enlarge the first number when reduce the plot size to avoid the error 'Error in plot.new() : figure margins too large' lwid = c(0.8, 4)) # enlarge the first number when reduce the plot size to avoid the error 'Error in plot.new() : figure margins too large' dev.off() png(paste0("DEGs_heatmap_", contrast, ".png"), width = 800, height = 1000) heatmap.2( datamat, Rowv = as.dendrogram(hr), Colv = FALSE, dendrogram = "row", col = bluered(75), scale = "row", trace = "none", density.info = "none", RowSideColors = mycol, margins = c(10, 15), # c(bottom, left) sepwidth = c(0, 0), labRow = labRow_pretty, labCol = labCol_pretty, cexRow = 1.3, cexCol = 1.8, srtCol = 15, lhei = c(0.01, 4), lwid = c(0.5, 4), key = FALSE # safer; add manual z-score key if you want (see note below) ) dev.off() # ------------------ Heatmap generation for three samples ---------------------- ## ============================================================ ## Three-condition DEGs heatmap from multiple pairwise contrasts ## Example contrasts: ## "WT_MH_4h_vs_WT_MH_2h", ## "WT_MH_18h_vs_WT_MH_2h", ## "WT_MH_18h_vs_WT_MH_4h" ## Output shows the union of DEGs across all contrasts and ## only the columns (samples) for the 3 conditions. ## ============================================================ ## -------- 0) User inputs ------------------------------------ contrasts <- c( "WT_MH_4h_vs_WT_MH_2h", "WT_MH_18h_vs_WT_MH_2h", "WT_MH_18h_vs_WT_MH_4h" ) contrasts <- c( "WT_TSB_4h_vs_WT_TSB_2h", "WT_TSB_18h_vs_WT_TSB_2h", "WT_TSB_18h_vs_WT_TSB_4h" ) contrasts <- c( "deltasbp_MH_4h_vs_deltasbp_MH_2h", "deltasbp_MH_18h_vs_deltasbp_MH_2h", "deltasbp_MH_18h_vs_deltasbp_MH_4h" ) contrasts <- c( "deltasbp_TSB_4h_vs_deltasbp_TSB_2h", "deltasbp_TSB_18h_vs_deltasbp_TSB_2h", "deltasbp_TSB_18h_vs_deltasbp_TSB_4h" ) ## Optionally force a condition display order (defaults to order of first appearance) cond_order <- c("WT_MH_2h","WT_MH_4h","WT_MH_18h") cond_order <- c("WT_TSB_2h","WT_TSB_4h","WT_TSB_18h") cond_order <- c("deltasbp_MH_2h","deltasbp_MH_4h","deltasbp_MH_18h") cond_order <- c("deltasbp_TSB_2h","deltasbp_TSB_4h","deltasbp_TSB_18h") #cond_order <- NULL ## -------- 1) Packages --------------------------------------- need <- c("gplots") to_install <- setdiff(need, rownames(installed.packages())) if (length(to_install)) install.packages(to_install, repos = "https://cloud.r-project.org") suppressPackageStartupMessages(library(gplots)) ## -------- 2) Helpers ---------------------------------------- read_ids_from_file <- function(path) { if (!file.exists(path)) stop("File not found: ", path) df <- tryCatch(read.table(path, header = TRUE, stringsAsFactors = FALSE, quote = "\"'", comment.char = ""), error = function(e) NULL) if (!is.null(df) && ncol(df) >= 1) { ids <- if ("Gene_Id" %in% names(df)) df[["Gene_Id"]] else df[[1]] } else { df2 <- read.table(path, header = FALSE, stringsAsFactors = FALSE, quote = "\"'", comment.char = "") ids <- df2[[1]] } ids <- trimws(gsub('"', "", ids)) ids[nzchar(ids)] } # From "A_vs_B" return c("A","B") split_contrast_groups <- function(x) { parts <- strsplit(x, "_vs_", fixed = TRUE)[[1]] if (length(parts) != 2L) stop("Contrast must be 'GroupA_vs_GroupB': ", x) parts } # Grep whole tag between start/end or underscores match_tags <- function(nms, tags) { pat <- paste0("(^|_)(?:", paste(tags, collapse = "|"), ")(_|$)") grepl(pat, nms, perl = TRUE) } # Pretty labels for columns (optional tweaks) prettify_col_labels <- function(x) { x <- gsub("^deltasbp", "\u0394sbp", x) # example from your earlier case x <- gsub("_", " ", x) x } # BREAK_LINE ## -------- 3) Build GOI (union across contrasts) ------------- up_files <- paste0(contrasts, "-up.id") down_files <- paste0(contrasts, "-down.id") GOI <- unique(unlist(c( lapply(up_files, read_ids_from_file), lapply(down_files, read_ids_from_file) ))) if (!length(GOI)) stop("No gene IDs found in any up/down .id files for the given contrasts.") ## -------- 4) Expression matrix (rld or vsd) ----------------- if (exists("rld")) { expr_all <- assay(rld) } else if (exists("vsd")) { expr_all <- assay(vsd) } else { stop("Neither 'rld' nor 'vsd' object is available in the environment.") } expr_all <- as.matrix(expr_all) present <- intersect(rownames(expr_all), GOI) if (!length(present)) stop("None of the GOI were found among the rownames of the expression matrix.") missing <- setdiff(GOI, present) if (length(missing)) message("Note: ", length(missing), " GOI not found and will be skipped.") ## -------- 5) Infer the THREE condition tags ----------------- pair_groups <- lapply(contrasts, split_contrast_groups) # list of c(A,B) cond_tags <- unique(unlist(pair_groups)) if (length(cond_tags) != 3L) { stop("Expected exactly three unique condition tags across the contrasts, got: ", paste(cond_tags, collapse = ", ")) } # If user provided an explicit order, use it; else keep first-appearance order if (!is.null(cond_order)) { if (!setequal(cond_order, cond_tags)) stop("cond_order must contain exactly these tags: ", paste(cond_tags, collapse = ", ")) cond_tags <- cond_order } #BREAK_LINE ## -------- 6) Subset columns to those 3 conditions ----------- # helper: does a name contain any of the tags? match_any_tag <- function(nms, tags) { pat <- paste0("(^|_)(?:", paste(tags, collapse = "|"), ")(_|$)") grepl(pat, nms, perl = TRUE) } # helper: return the specific tag that a single name matches detect_tag <- function(nm, tags) { hits <- vapply(tags, function(t) grepl(paste0("(^|_)", t, "(_|$)"), nm, perl = TRUE), logical(1)) if (!any(hits)) NA_character_ else tags[which(hits)[1]] } keep_cols <- match_any_tag(colnames(expr_all), cond_tags) if (!any(keep_cols)) { stop("No columns matched any of the three condition tags: ", paste(cond_tags, collapse = ", ")) } sub_idx <- which(keep_cols) sub_colnames <- colnames(expr_all)[sub_idx] # find the tag for each kept column (this is the part that was wrong before) cond_for_col <- vapply(sub_colnames, detect_tag, character(1), tags = cond_tags) # rank columns by your desired condition order, then by name within each condition cond_rank <- match(cond_for_col, cond_tags) ord <- order(cond_rank, sub_colnames) expr_sub <- expr_all[present, sub_idx, drop = FALSE][, ord, drop = FALSE] ## -------- 7) Remove constant/NA rows ------------------------ row_ok <- apply(expr_sub, 1, function(x) is.finite(sum(x)) && var(x, na.rm = TRUE) > 0) if (any(!row_ok)) message("Removing ", sum(!row_ok), " constant/NA rows.") datamat <- expr_sub[row_ok, , drop = FALSE] ## -------- 8) Labels ---------------------------------------- labRow_pretty <- sub("^gene-", "", rownames(datamat)) labCol_pretty <- prettify_col_labels(colnames(datamat)) ## -------- 9) Clustering (rows) ------------------------------ hr <- hclust(as.dist(1 - cor(t(datamat), method = "pearson")), method = "complete") # Color row-side groups by cutting the dendrogram mycl <- cutree(hr, h = max(hr$height) / 1.3) palette_base <- c("yellow","blue","orange","magenta","cyan","red","green","maroon", "lightblue","pink","purple","lightcyan","salmon","lightgreen") mycol <- palette_base[(as.vector(mycl) - 1) %% length(palette_base) + 1] ## -------- 10) Save the matrix used -------------------------- out_tag <- paste(cond_tags, collapse = "_") write.csv(as.data.frame(datamat), file = paste0("DEGs_heatmap_expression_data_", out_tag, ".txt"), quote = FALSE) ## -------- 11) Plot heatmap ---------------------------------- png(paste0("DEGs_heatmap_", out_tag, ".png"), width = 1000, height = 5000) heatmap.2( datamat, Rowv = as.dendrogram(hr), Colv = FALSE, dendrogram = "row", col = bluered(75), scale = "row", trace = "none", density.info = "none", RowSideColors = mycol, margins = c(10, 15), # c(bottom, left) sepwidth = c(0, 0), labRow = labRow_pretty, labCol = labCol_pretty, cexRow = 1.3, cexCol = 1.8, srtCol = 15, lhei = c(0.01, 4), lwid = c(0.5, 4), key = FALSE # safer; add manual z-score key if you want (see note below) ) dev.off() mv DEGs_heatmap_WT_MH_2h_WT_MH_4h_WT_MH_18h.png DEGs_heatmap_WT_MH.png mv DEGs_heatmap_WT_TSB_2h_WT_TSB_4h_WT_TSB_18h.png DEGs_heatmap_WT_TSB.png mv DEGs_heatmap_deltasbp_MH_2h_deltasbp_MH_4h_deltasbp_MH_18h.png DEGs_heatmap_deltasbp_MH.png mv DEGs_heatmap_deltasbp_TSB_2h_deltasbp_TSB_4h_deltasbp_TSB_18h.png DEGs_heatmap_deltasbp_TSB.png # ------------------ Heatmap generation for three samples END ---------------------- # ==== (NOT_USED) Ultra-robust heatmap.2 plotter with many attempts ==== # Inputs: # mat : numeric matrix (genes x samples) # hr : hclust for rows (or TRUE/FALSE) # row_colors : vector of RowSideColors of length nrow(mat) or NULL # labRow : character vector of row labels (display only) # labCol : character vector of col labels (display only) # outfile : output PNG path # base_res : DPI for PNG (default 150) # ==== Slide-tuned heatmap.2 plotter (moderate size, larger fonts, 45° labels) ==== safe_heatmap2 <- function(mat, hr, row_colors, labRow, labCol, outfile, base_res = 150) { stopifnot(is.matrix(mat)) nr <- nrow(mat); nc <- ncol(mat) # Target slide size & sensible caps #target_w <- 2400; target_h <- 1600 #max_w <- 3000; max_h <- 2000 target_w <- 800; target_h <- 2000 max_w <- 1500; max_h <- 1500 # Label stats max_row_chars <- if (length(labRow)) max(nchar(labRow), na.rm = TRUE) else 1 max_col_chars <- if (length(labCol)) max(nchar(labCol), na.rm = TRUE) else 1 #add_attempt(target_w, target_h, 0.90, 1.00, 45, NULL, TRUE, TRUE, TRUE) attempts <- list() add_attempt <- function(w, h, cr, cc, rot, mar = NULL, key = TRUE, showR = TRUE, showC = TRUE, trunc_rows = 0) { attempts[[length(attempts) + 1]] <<- list( w = w, h = h, cr = cr, cc = cc, rot = rot, mar = mar, key = key, showR = showR, showC = showC, trunc_rows = trunc_rows ) } # Note that if the key is FALSE, all works, if the key is TRUE, none works! # 1) Preferred look: moderate size, biggish fonts, 45° labels add_attempt(target_w, target_h, 0.90, 1.00, 30, c(2,1), TRUE, TRUE, TRUE) # 2) Same, explicit margins computed later add_attempt(target_w, target_h, 0.85, 0.95, 45, c(10,15), TRUE, TRUE, TRUE) # 3) Slightly bigger canvas add_attempt(min(target_w+300, max_w), min(target_h+200, max_h), 0.85, 0.95, 30, c(10,15), TRUE, TRUE, TRUE) # 4) Make margins more generous (in lines) add_attempt(min(target_w+300, max_w), min(target_h+200, max_h), 0.80, 0.90, 30, c(10,14), FALSE, TRUE, TRUE) # 5) Reduce rotation to 30 if still tight add_attempt(min(target_w+300, max_w), min(target_h+200, max_h), 0.80, 0.90, 30, c(8,12), FALSE, TRUE, TRUE) # 6) Final fallback: keep fonts reasonable, 0° labels, slightly bigger margins add_attempt(min(target_w+500, max_w), min(target_h+300, max_h), 0.80, 0.90, 45, c(8,12), FALSE, TRUE, TRUE) # 7) Last resort: truncate long row labels (keeps readability) if (max_row_chars > 20) { add_attempt(min(target_w+500, max_w), min(target_h+300, max_h), 0.80, 0.90, 30, c(8,12), FALSE, TRUE, TRUE, trunc_rows = 18) } for (i in seq_along(attempts)) { a <- attempts[[i]] # Compute margins if not provided if (is.null(a$mar)) { col_margin <- if (a$showC) { if (a$rot > 0) max(6, ceiling(0.45 * max_col_chars * max(a$cc, 0.8))) else max(5, ceiling(0.22 * max_col_chars * max(a$cc, 0.8))) } else 4 row_margin <- if (a$showR) max(6, ceiling(0.55 * max_row_chars * max(a$cr, 0.8))) else 4 mar <- c(col_margin, row_margin) } else { mar <- a$mar } # Prepare labels for this attempt lr <- if (a$showR) labRow else rep("", nr) if (a$trunc_rows > 0 && a$showR) { lr <- ifelse(nchar(lr) > a$trunc_rows, paste0(substr(lr, 1, a$trunc_rows), "…"), lr) } lc <- if (a$showC) labCol else rep("", nc) # Close any open device if (dev.cur() != 1) try(dev.off(), silent = TRUE) ok <- FALSE try({ png(outfile, width = ceiling(a$w), height = ceiling(a$h), res = base_res) gplots::heatmap.2( mat, Rowv = as.dendrogram(hr), Colv = FALSE, dendrogram = "row", col = gplots::bluered(75), scale = "row", trace = "none", density.info = "none", RowSideColors = row_colors, key = a$key, margins = mar, # c(col, row) in lines sepwidth = c(0, 0), labRow = lr, labCol = lc, cexRow = a$cr, cexCol = a$cc, srtCol = a$rot, lhei = c(0.1, 4), lwid = c(0.1, 4) ) dev.off() ok <- TRUE }, silent = TRUE) if (ok) { message(sprintf( "✓ Heatmap saved: %s (attempt %d) size=%dx%d margins=c(%d,%d) cexRow=%.2f cexCol=%.2f srtCol=%d", outfile, i, ceiling(a$w), ceiling(a$h), mar[1], mar[2], a$cr, a$cc, a$rot )) return(invisible(TRUE)) } else { if (dev.cur() != 1) try(dev.off(), silent = TRUE) message(sprintf("Attempt %d failed; retrying...", i)) } } stop("Failed to draw heatmap after tuned attempts. Consider ComplexHeatmap if this persists.") } safe_heatmap2( mat = datamat, hr = hr, row_colors = mycol, labRow = labRow_pretty, # row labels WITHOUT "gene-" labCol = labCol_pretty, # col labels with Δsbp + spaces outfile = paste0("DEGs_heatmap_", contrast, ".png"), #base_res = 150 ) # -- (OLD CODE) DEGs_heatmap_deltasbp_TSB_2h_vs_WT_TSB_2h -- cat deltasbp_TSB_2h_vs_WT_TSB_2h-up.id deltasbp_TSB_2h_vs_WT_TSB_2h-down.id | sort -u > ids #add Gene_Id in the first line, delete the "" #Note that using GeneID as index, rather than GeneName, since .txt contains only GeneID. GOI <- read.csv("ids")$Gene_Id RNASeq.NoCellLine <- assay(rld) #install.packages("gplots") library("gplots") #clustering methods: "ward.D", "ward.D2", "single", "complete", "average" (= UPGMA), "mcquitty" (= WPGMA), "median" (= WPGMC) or "centroid" (= UPGMC). pearson or spearman datamat = RNASeq.NoCellLine[GOI, ] #datamat = RNASeq.NoCellLine write.csv(as.data.frame(datamat), file ="DEGs_heatmap_expression_data.txt") constant_rows <- apply(datamat, 1, function(row) var(row) == 0) if(any(constant_rows)) { cat("Removing", sum(constant_rows), "constant rows.\n") datamat <- datamat[!constant_rows, ] } hr <- hclust(as.dist(1-cor(t(datamat), method="pearson")), method="complete") hc <- hclust(as.dist(1-cor(datamat, method="spearman")), method="complete") mycl = cutree(hr, h=max(hr$height)/1.1) mycol = c("YELLOW", "BLUE", "ORANGE", "MAGENTA", "CYAN", "RED", "GREEN", "MAROON", "LIGHTBLUE", "PINK", "MAGENTA", "LIGHTCYAN", "LIGHTRED", "LIGHTGREEN"); mycol = mycol[as.vector(mycl)] png("DEGs_heatmap_deltasbp_TSB_2h_vs_WT_TSB_2h.png", width=1200, height=2000) heatmap.2(datamat, Rowv = as.dendrogram(hr), col = bluered(75), scale = "row", RowSideColors = mycol, trace = "none", margin = c(10, 15), # bottom, left sepwidth = c(0, 0), dendrogram = 'row', Colv = 'false', density.info = 'none', labRow = rownames(datamat), cexRow = 1.5, cexCol = 1.5, srtCol = 35, lhei = c(0.2, 4), # reduce top space (was 1 or more) lwid = c(0.4, 4)) # reduce left space (was 1 or more) dev.off() # -------------- Cluster members ---------------- write.csv(names(subset(mycl, mycl == '1')),file='cluster1_YELLOW.txt') write.csv(names(subset(mycl, mycl == '2')),file='cluster2_DARKBLUE.txt') write.csv(names(subset(mycl, mycl == '3')),file='cluster3_DARKORANGE.txt') write.csv(names(subset(mycl, mycl == '4')),file='cluster4_DARKMAGENTA.txt') write.csv(names(subset(mycl, mycl == '5')),file='cluster5_DARKCYAN.txt') #~/Tools/csv2xls-0.4/csv_to_xls.py cluster*.txt -d',' -o DEGs_heatmap_cluster_members.xls #~/Tools/csv2xls-0.4/csv_to_xls.py DEGs_heatmap_expression_data.txt -d',' -o DEGs_heatmap_expression_data.xls; #### (NOT_WORKING) cluster members (adding annotations, note that it does not work for the bacteria, since it is not model-speices and we cannot use mart=ensembl) ##### subset_1<-names(subset(mycl, mycl == '1')) data <- as.data.frame(datamat[rownames(datamat) %in% subset_1, ]) #2575 subset_2<-names(subset(mycl, mycl == '2')) data <- as.data.frame(datamat[rownames(datamat) %in% subset_2, ]) #1855 subset_3<-names(subset(mycl, mycl == '3')) data <- as.data.frame(datamat[rownames(datamat) %in% subset_3, ]) #217 subset_4<-names(subset(mycl, mycl == '4')) data <- as.data.frame(datamat[rownames(datamat) %in% subset_4, ]) # subset_5<-names(subset(mycl, mycl == '5')) data <- as.data.frame(datamat[rownames(datamat) %in% subset_5, ]) # # Initialize an empty data frame for the annotated data annotated_data <- data.frame() # Determine total number of genes total_genes <- length(rownames(data)) # Loop through each gene to annotate for (i in 1:total_genes) { gene <- rownames(data)[i] result <- getBM(attributes = c('ensembl_gene_id', 'external_gene_name', 'gene_biotype', 'entrezgene_id', 'chromosome_name', 'start_position', 'end_position', 'strand', 'description'), filters = 'ensembl_gene_id', values = gene, mart = ensembl) # If multiple rows are returned, take the first one if (nrow(result) > 1) { result <- result[1, ] } # Check if the result is empty if (nrow(result) == 0) { result <- data.frame(ensembl_gene_id = gene, external_gene_name = NA, gene_biotype = NA, entrezgene_id = NA, chromosome_name = NA, start_position = NA, end_position = NA, strand = NA, description = NA) } # Transpose expression values expression_values <- t(data.frame(t(data[gene, ]))) colnames(expression_values) <- colnames(data) # Combine gene information and expression data combined_result <- cbind(result, expression_values) # Append to the final dataframe annotated_data <- rbind(annotated_data, combined_result) # Print progress every 100 genes if (i %% 100 == 0) { cat(sprintf("Processed gene %d out of %d\n", i, total_genes)) } } # Save the annotated data to a new CSV file write.csv(annotated_data, "cluster1_YELLOW.csv", row.names=FALSE) write.csv(annotated_data, "cluster2_DARKBLUE.csv", row.names=FALSE) write.csv(annotated_data, "cluster3_DARKORANGE.csv", row.names=FALSE) write.csv(annotated_data, "cluster4_DARKMAGENTA.csv", row.names=FALSE) write.csv(annotated_data, "cluster5_DARKCYAN.csv", row.names=FALSE) #~/Tools/csv2xls-0.4/csv_to_xls.py cluster*.csv -d',' -o DEGs_heatmap_clusters.xls
KEGG and GO annotations in non-model organisms
https://www.biobam.com/functional-analysis/

-
Assign KEGG and GO Terms (see diagram above)
Since your organism is non-model, standard R databases (org.Hs.eg.db, etc.) won’t work. You’ll need to manually retrieve KEGG and GO annotations. Option 1 (KEGG Terms): EggNog based on orthology and phylogenies EggNOG-mapper assigns both KEGG Orthology (KO) IDs and GO terms. Install EggNOG-mapper: mamba create -n eggnog_env python=3.8 eggnog-mapper -c conda-forge -c bioconda #eggnog-mapper_2.1.12 mamba activate eggnog_env Run annotation: #diamond makedb --in eggnog6.prots.faa -d eggnog_proteins.dmnd mkdir /home/jhuang/mambaforge/envs/eggnog_env/lib/python3.8/site-packages/data/ download_eggnog_data.py --dbname eggnog.db -y --data_dir /home/jhuang/mambaforge/envs/eggnog_env/lib/python3.8/site-packages/data/ #NOT_WORKING: emapper.py -i CP020463_gene.fasta -o eggnog_dmnd_out --cpu 60 -m diamond[hmmer,mmseqs] --dmnd_db /home/jhuang/REFs/eggnog_data/data/eggnog_proteins.dmnd #Download the protein sequences from Genbank mv ~/Downloads/sequence\ \(3\).txt CP020463_protein_.fasta python ~/Scripts/update_fasta_header.py CP020463_protein_.fasta CP020463_protein.fasta emapper.py -i CP020463_protein.fasta -o eggnog_out --cpu 60 #--resume #----> result annotations.tsv: Contains KEGG, GO, and other functional annotations. #----> 470.IX87_14445: * 470 likely refers to the organism or strain (e.g., Acinetobacter baumannii ATCC 19606 or another related strain). * IX87_14445 would refer to a specific gene or protein within that genome. Extract KEGG KO IDs from annotations.emapper.annotations. Option 2 (GO Terms from 'Blast2GO 5 Basic', saved in blast2go_annot.annot): Using Blast/Diamond + Blast2GO_GUI based on sequence alignment + GO mapping * jhuang@WS-2290C:~/DATA/Data_Michelle_RNAseq_2025$ ~/Tools/Blast2GO/Blast2GO_Launcher setting the workspace "mkdir ~/b2gWorkspace_Michelle_RNAseq_2025"; cp /mnt/md1/DATA/Data_Michelle_RNAseq_2025/results/star_salmon/degenes/CP020463_protein.fasta ~/b2gWorkspace_Michelle_RNAseq_2025 * 'Load protein sequences' (Tags: NONE, generated columns: Nr, SeqName) by choosing the file CP020463_protein.fasta as input --> * Buttons 'blast' at the NCBI (Parameters: blastp, nr, ...) (Tags: BLASTED, generated columns: Description, Length, #Hits, e-Value, sim mean), QBlast finished with warnings! Blasted Sequences: 2084 Sequences without results: 105 Check the Job log for details and try to submit again. Restarting QBlast may result in additional results, depending on the error type. "Blast (CP020463_protein) Done" * Button 'mapping' (Tags: MAPPED, generated columns: #GO, GO IDs, GO Names), "Mapping finished - Please proceed now to annotation." "Mapping (CP020463_protein) Done" "Mapping finished - Please proceed now to annotation." * Button 'annot' (Tags: ANNOTATED, generated columns: Enzyme Codes, Enzyme Names), "Annotation finished." * Used parameter 'Annotation CutOff': The Blast2GO Annotation Rule seeks to find the most specific GO annotations with a certain level of reliability. An annotation score is calculated for each candidate GO which is composed by the sequence similarity of the Blast Hit, the evidence code of the source GO and the position of the particular GO in the Gene Ontology hierarchy. This annotation score cutoff select the most specific GO term for a given GO branch which lies above this value. * Used parameter 'GO Weight' is a value which is added to Annotation Score of a more general/abstract Gene Ontology term for each of its more specific, original source GO terms. In this case, more general GO terms which summarise many original source terms (those ones directly associated to the Blast Hits) will have a higher Annotation Score. "Annotation (CP020463_protein) Done" "Annotation finished." or blast2go_cli_v1.5.1 (NOT_USED) #https://help.biobam.com/space/BCD/2250407989/Installation #see ~/Scripts/blast2go_pipeline.sh Option 3 (GO Terms from 'Blast2GO 5 Basic', saved in blast2go_annot.annot2): Interpro based protein families / domains --> Button interpro * Button 'interpro' (Tags: INTERPRO, generated columns: InterPro IDs, InterPro GO IDs, InterPro GO Names) --> "InterProScan Finished - You can now merge the obtained GO Annotations." "InterProScan (CP020463_protein) Done" "InterProScan Finished - You can now merge the obtained GO Annotations." MERGE the results of InterPro GO IDs (Option 3) to GO IDs (Option 2) and generate final GO IDs * Button 'interpro'/'Merge InterProScan GOs to Annotation' --> "Merge (add and validate) all GO terms retrieved via InterProScan to the already existing GO annotation." "Merge InterProScan GOs to Annotation (CP020463_protein) Done" "Finished merging GO terms from InterPro with annotations." "Maybe you want to run ANNEX (Annotation Augmentation)." #* Button 'annot'/'ANNEX' --> "ANNEX finished. Maybe you want to do the next step: Enzyme Code Mapping." File -> Export -> Export Annotations -> Export Annotations (.annot, custom, etc.) #~/b2gWorkspace_Michelle_RNAseq_2025/blast2go_annot.annot is generated! #-- before merging (blast2go_annot.annot) -- #H0N29_18790 GO:0004842 ankyrin repeat domain-containing protein #H0N29_18790 GO:0085020 #-- after merging (blast2go_annot.annot2) --> #H0N29_18790 GO:0031436 ankyrin repeat domain-containing protein #H0N29_18790 GO:0070531 #H0N29_18790 GO:0004842 #H0N29_18790 GO:0005515 #H0N29_18790 GO:0085020 cp blast2go_annot.annot blast2go_annot.annot2 Option 4 (NOT_USED): RFAM for non-colding RNA Option 5 (NOT_USED): PSORTb for subcellular localizations Option 6 (NOT_USED): KAAS (KEGG Automatic Annotation Server) * Go to KAAS * Upload your FASTA file. * Select an appropriate gene set. * Download the KO assignments. -
Find the Closest KEGG Organism Code (NOT_USED)
Since your species isn't directly in KEGG, use a closely related organism. * Check available KEGG organisms: library(clusterProfiler) library(KEGGREST) kegg_organisms <- keggList("organism") Pick the closest relative (e.g., zebrafish "dre" for fish, Arabidopsis "ath" for plants). # Search for Acinetobacter in the list grep("Acinetobacter", kegg_organisms, ignore.case = TRUE, value = TRUE) # Gammaproteobacteria #Extract KO IDs from the eggnog results for "Acinetobacter baumannii strain ATCC 19606" -
Find the Closest KEGG Organism for a Non-Model Species (NOT_USED)
If your organism is not in KEGG, search for the closest relative: grep("fish", kegg_organisms, ignore.case = TRUE, value = TRUE) # Example search For KEGG pathway enrichment in non-model species, use "ko" instead of a species code (the code has been intergrated in the point 4): kegg_enrich <- enrichKEGG(gene = gene_list, organism = "ko") # "ko" = KEGG Orthology -
Perform KEGG and GO Enrichment in R (under dir ~/DATA/Data_Tam_RNAseq_2025_subMIC_exposure_ATCC19606/results/star_salmon/degenes)
#BiocManager::install("GO.db") #BiocManager::install("AnnotationDbi") # Load required libraries library(openxlsx) # For Excel file handling library(dplyr) # For data manipulation library(tidyr) library(stringr) library(clusterProfiler) # For KEGG and GO enrichment analysis #library(org.Hs.eg.db) # Replace with appropriate organism database library(GO.db) library(AnnotationDbi) setwd("~/DATA/Data_Tam_RNAseq_2025_subMIC_exposure_ATCC19606/results/star_salmon/degenes") # PREPARING go_terms and ec_terms: annot_* file: cut -f1-2 -d$'\t' blast2go_annot.annot2 > blast2go_annot.annot2_ # PREPARING eggnog_out.emapper.annotations.txt from eggnog_out.emapper.annotations by removing ## lines and renaming #query to query #(plot-numpy1) jhuang@WS-2290C:~/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606$ diff eggnog_out.emapper.annotations eggnog_out.emapper.annotations.txt #1,5c1 #< ## Thu Jan 30 16:34:52 2025 #< ## emapper-2.1.12 #< ## /home/jhuang/mambaforge/envs/eggnog_env/bin/emapper.py -i CP059040_protein.fasta -o eggnog_out --cpu 60 #< ## #< #query seed_ortholog evalue score eggNOG_OGs max_annot_lvl COG_category Description Preferred_name GOs EC KEGG_ko KEGG_Pathway KEGG_Module KEGG_Reaction KEGG_rclass BRITE KEGG_TC CAZy BiGG_Reaction PFAMs #--- #> query seed_ortholog evalue score eggNOG_OGs max_annot_lvl COG_category Description Preferred_name GOs EC KEGG_ko KEGG_Pathway KEGG_Module KEGG_Reaction KEGG_rclass BRITE KEGG_TC CAZy BiGG_Reaction PFAMs #3620,3622d3615 #< ## 3614 queries scanned #< ## Total time (seconds): 8.176708459854126 # Step 1: Load the blast2go annotation file with a check for missing columns annot_df <- read.table("/home/jhuang/b2gWorkspace_Tam_RNAseq_2024/blast2go_annot.annot2_", header = FALSE, sep = "\t", stringsAsFactors = FALSE, fill = TRUE) # If the structure is inconsistent, we can make sure there are exactly 3 columns: colnames(annot_df) <- c("GeneID", "Term") # Step 2: Filter and aggregate GO and EC terms as before go_terms <- annot_df %>% filter(grepl("^GO:", Term)) %>% group_by(GeneID) %>% summarize(GOs = paste(Term, collapse = ","), .groups = "drop") ec_terms <- annot_df %>% filter(grepl("^EC:", Term)) %>% group_by(GeneID) %>% summarize(EC = paste(Term, collapse = ","), .groups = "drop") # Key Improvements: # * Looped processing of all 6 input files to avoid redundancy. # * Robust handling of empty KEGG and GO enrichment results to prevent contamination of results between iterations. # * File-safe output: Each dataset creates a separate Excel workbook with enriched sheets only if data exists. # * Error handling for GO term descriptions via tryCatch. # * Improved clarity and modular structure for easier maintenance and future additions. #file_name = "deltasbp_TSB_2h_vs_WT_TSB_2h-all.csv" # ---------------------- Generated DEG(Annotated)_KEGG_GO_* ----------------------- suppressPackageStartupMessages({ library(readr) library(dplyr) library(stringr) library(tidyr) library(openxlsx) library(clusterProfiler) library(AnnotationDbi) library(GO.db) }) # ---- PARAMETERS ---- PADJ_CUT <- 5e-2 LFC_CUT <- 2 # Your emapper annotations (with columns: query, GOs, EC, KEGG_ko, KEGG_Pathway, KEGG_Module, ... ) emapper_path <- "~/DATA/Data_Tam_RNAseq_2024_AUM_MHB_Urine_ATCC19606/eggnog_out.emapper.annotations.txt" # Input files (you can add/remove here) input_files <- c( "deltaadeIJ_none_17_vs_WT_none_17-all.csv", #up 11, down 3 vs. (10,4) "deltaadeIJ_none_24_vs_WT_none_24-all.csv", #up 0, down 2 vs. (0,2) "deltaadeIJ_one_17_vs_WT_one_17-all.csv", #up 238, down 90 vs. (239,89) --> height 2600 "deltaadeIJ_one_24_vs_WT_one_24-all.csv", #up 83, down 64 vs. (64,71) --> height 1600 "deltaadeIJ_two_17_vs_WT_two_17-all.csv", #up 74, down 14 vs. (75,9) --> height 1000 "deltaadeIJ_two_24_vs_WT_two_24-all.csv", #up 1, down 3 vs. (3,3) "WT_none_24_vs_WT_none_17-all.csv", #(up 10, down 2) "WT_one_24_vs_WT_one_17-all.csv", #(up 97, down 3) "WT_two_24_vs_WT_two_17-all.csv", #(up 12, down 1) "deltaadeIJ_two_24_vs_deltaadeIJ_two_17-all.csv", #(up 8, down 51) "deltaadeIJ_one_24_vs_deltaadeIJ_one_17-all.csv", #(up 0, down 10) "deltaadeIJ_none_24_vs_deltaadeIJ_none_17-all.csv" #(up 0, down 0) ) # ---- HELPERS ---- # Robust reader (CSV first, then TSV) read_table_any <- function(path) { tb <- tryCatch(readr::read_csv(path, show_col_types = FALSE), error = function(e) tryCatch(readr::read_tsv(path, col_types = cols()), error = function(e2) NULL)) tb } # Return a nice Excel-safe base name xlsx_name_from_file <- function(path) { base <- tools::file_path_sans_ext(basename(path)) paste0("DEG_KEGG_GO_", base, ".xlsx") } # KEGG expand helper: replace K-numbers with GeneIDs using mapping from the same result table expand_kegg_geneIDs <- function(kegg_res, mapping_tbl) { if (is.null(kegg_res) || nrow(as.data.frame(kegg_res)) == 0) return(data.frame()) kdf <- as.data.frame(kegg_res) if (!"geneID" %in% names(kdf)) return(kdf) # mapping_tbl: columns KEGG_ko (possibly multiple separated by commas) and GeneID map_clean <- mapping_tbl %>% dplyr::select(KEGG_ko, GeneID) %>% filter(!is.na(KEGG_ko), KEGG_ko != "-") %>% mutate(KEGG_ko = str_remove_all(KEGG_ko, "ko:")) %>% tidyr::separate_rows(KEGG_ko, sep = ",") %>% distinct() if (!nrow(map_clean)) { return(kdf) } expanded <- kdf %>% tidyr::separate_rows(geneID, sep = "/") %>% dplyr::left_join(map_clean, by = c("geneID" = "KEGG_ko"), relationship = "many-to-many") %>% distinct() %>% dplyr::group_by(ID) %>% dplyr::summarise(across(everything(), ~ paste(unique(na.omit(.)), collapse = "/")), .groups = "drop") kdf %>% dplyr::select(-geneID) %>% dplyr::left_join(expanded %>% dplyr::select(ID, GeneID), by = "ID") %>% dplyr::rename(geneID = GeneID) } # ---- LOAD emapper annotations ---- eggnog_data <- read.delim(emapper_path, header = TRUE, sep = "\t", quote = "", check.names = FALSE) # Ensure character columns for joins eggnog_data$query <- as.character(eggnog_data$query) eggnog_data$GOs <- as.character(eggnog_data$GOs) eggnog_data$EC <- as.character(eggnog_data$EC) eggnog_data$KEGG_ko <- as.character(eggnog_data$KEGG_ko) # ---- MAIN LOOP ---- for (f in input_files) { if (!file.exists(f)) { message("Missing: ", f); next } message("Processing: ", f) res <- read_table_any(f) if (is.null(res) || nrow(res) == 0) { message("Empty/unreadable: ", f); next } # Coerce expected columns if present if ("padj" %in% names(res)) res$padj <- suppressWarnings(as.numeric(res$padj)) if ("log2FoldChange" %in% names(res)) res$log2FoldChange <- suppressWarnings(as.numeric(res$log2FoldChange)) # Ensure GeneID & GeneName exist if (!"GeneID" %in% names(res)) { # Try to infer from a generic 'gene' column if ("gene" %in% names(res)) res$GeneID <- as.character(res$gene) else res$GeneID <- NA_character_ } if (!"GeneName" %in% names(res)) res$GeneName <- NA_character_ # Fill missing GeneName from GeneID (drop "gene-") res$GeneName <- ifelse(is.na(res$GeneName) | res$GeneName == "", gsub("^gene-", "", as.character(res$GeneID)), as.character(res$GeneName)) # De-duplicate by GeneName, keep smallest padj if (!"padj" %in% names(res)) res$padj <- NA_real_ res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() %>% as.data.frame() # Sort by padj asc, then log2FC desc if (!"log2FoldChange" %in% names(res)) res$log2FoldChange <- NA_real_ res <- res[order(res$padj, -res$log2FoldChange), , drop = FALSE] # Join emapper (strip "gene-" from GeneID to match emapper 'query') res$GeneID_plain <- gsub("^gene-", "", res$GeneID) res_ann <- res %>% left_join(eggnog_data, by = c("GeneID_plain" = "query")) # --- Split by UP/DOWN using your volcano cutoffs --- up_regulated <- res_ann %>% filter(!is.na(padj), padj < PADJ_CUT, log2FoldChange > LFC_CUT) down_regulated <- res_ann %>% filter(!is.na(padj), padj < PADJ_CUT, log2FoldChange < -LFC_CUT) # --- KEGG enrichment (using K numbers in KEGG_ko) --- # Prepare KO lists (remove "ko:" if present) k_up <- up_regulated$KEGG_ko; k_up <- k_up[!is.na(k_up)] k_dn <- down_regulated$KEGG_ko; k_dn <- k_dn[!is.na(k_dn)] k_up <- gsub("ko:", "", k_up); k_dn <- gsub("ko:", "", k_dn) # BREAK_LINE kegg_up <- tryCatch(enrichKEGG(gene = k_up, organism = "ko"), error = function(e) NULL) kegg_down <- tryCatch(enrichKEGG(gene = k_dn, organism = "ko"), error = function(e) NULL) # Convert KEGG K-numbers to your GeneIDs (using mapping from the same result set) kegg_up_df <- expand_kegg_geneIDs(kegg_up, up_regulated) kegg_down_df <- expand_kegg_geneIDs(kegg_down, down_regulated) # --- GO enrichment (custom TERM2GENE built from emapper GOs) --- # Background gene set = all genes in this comparison background_genes <- unique(res_ann$GeneID_plain) # TERM2GENE table (GO -> GeneID_plain) go_annotation <- res_ann %>% dplyr::select(GeneID_plain, GOs) %>% mutate(GOs = ifelse(is.na(GOs), "", GOs)) %>% tidyr::separate_rows(GOs, sep = ",") %>% filter(GOs != "") %>% dplyr::select(GOs, GeneID_plain) %>% distinct() # Gene lists for GO enricher go_list_up <- unique(up_regulated$GeneID_plain) go_list_down <- unique(down_regulated$GeneID_plain) go_up <- tryCatch( enricher(gene = go_list_up, TERM2GENE = go_annotation, pvalueCutoff = 0.05, pAdjustMethod = "BH", universe = background_genes), error = function(e) NULL ) go_down <- tryCatch( enricher(gene = go_list_down, TERM2GENE = go_annotation, pvalueCutoff = 0.05, pAdjustMethod = "BH", universe = background_genes), error = function(e) NULL ) go_up_df <- if (!is.null(go_up)) as.data.frame(go_up) else data.frame() go_down_df <- if (!is.null(go_down)) as.data.frame(go_down) else data.frame() # Add GO term descriptions via GO.db (best-effort) add_go_term_desc <- function(df) { if (!nrow(df) || !"ID" %in% names(df)) return(df) df$Description <- sapply(df$ID, function(go_id) { term <- tryCatch(AnnotationDbi::select(GO.db, keys = go_id, columns = "TERM", keytype = "GOID"), error = function(e) NULL) if (!is.null(term) && nrow(term)) term$TERM[1] else NA_character_ }) df } go_up_df <- add_go_term_desc(go_up_df) go_down_df <- add_go_term_desc(go_down_df) # ---- Write Excel workbook ---- out_xlsx <- xlsx_name_from_file(f) wb <- createWorkbook() addWorksheet(wb, "Complete") writeData(wb, "Complete", res_ann) addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) addWorksheet(wb, "KEGG_Enrichment_Up") writeData(wb, "KEGG_Enrichment_Up", kegg_up_df) addWorksheet(wb, "KEGG_Enrichment_Down") writeData(wb, "KEGG_Enrichment_Down", kegg_down_df) addWorksheet(wb, "GO_Enrichment_Up") writeData(wb, "GO_Enrichment_Up", go_up_df) addWorksheet(wb, "GO_Enrichment_Down") writeData(wb, "GO_Enrichment_Down", go_down_df) saveWorkbook(wb, out_xlsx, overwrite = TRUE) message("Saved: ", out_xlsx) } # -------------------------------- OLD_CODE not automatized with loop ---------------------------- # Load the results res <- read.csv("deltasbp_TSB_2h_vs_WT_TSB_2h-all.csv") res <- read.csv("deltasbp_TSB_4h_vs_WT_TSB_4h-all.csv") res <- read.csv("deltasbp_TSB_18h_vs_WT_TSB_18h-all.csv") res <- read.csv("deltasbp_MH_2h_vs_WT_MH_2h-all.csv") res <- read.csv("deltasbp_MH_4h_vs_WT_MH_4h-all.csv") res <- read.csv("deltasbp_MH_18h_vs_WT_MH_18h-all.csv") res <- read.csv("WT_MH_4h_vs_WT_MH_2h-all.csv") res <- read.csv("WT_MH_18h_vs_WT_MH_2h-all.csv") res <- read.csv("WT_MH_18h_vs_WT_MH_4h-all.csv") res <- read.csv("WT_TSB_4h_vs_WT_TSB_2h-all.csv") res <- read.csv("WT_TSB_18h_vs_WT_TSB_2h-all.csv") res <- read.csv("WT_TSB_18h_vs_WT_TSB_4h-all.csv") res <- read.csv("deltasbp_MH_4h_vs_deltasbp_MH_2h-all.csv") res <- read.csv("deltasbp_MH_18h_vs_deltasbp_MH_2h-all.csv") res <- read.csv("deltasbp_MH_18h_vs_deltasbp_MH_4h-all.csv") res <- read.csv("deltasbp_TSB_4h_vs_deltasbp_TSB_2h-all.csv") res <- read.csv("deltasbp_TSB_18h_vs_deltasbp_TSB_2h-all.csv") res <- read.csv("deltasbp_TSB_18h_vs_deltasbp_TSB_4h-all.csv") # Replace empty GeneName with modified GeneID res$GeneName <- ifelse( res$GeneName == "" | is.na(res$GeneName), gsub("gene-", "", res$GeneID), res$GeneName ) # Remove duplicated genes by selecting the gene with the smallest padj duplicated_genes <- res[duplicated(res$GeneName), "GeneName"] res <- res %>% group_by(GeneName) %>% slice_min(padj, with_ties = FALSE) %>% ungroup() res <- as.data.frame(res) # Sort res first by padj (ascending) and then by log2FoldChange (descending) res <- res[order(res$padj, -res$log2FoldChange), ] # Read eggnog annotations eggnog_data <- read.delim("~/DATA/Data_Michelle_RNAseq_2025/eggnog_out.emapper.annotations.txt", header = TRUE, sep = "\t") # Remove the "gene-" prefix from GeneID in res to match eggnog 'query' format res$GeneID <- gsub("gene-", "", res$GeneID) # Merge eggnog data with res based on GeneID res <- res %>% left_join(eggnog_data, by = c("GeneID" = "query")) # Merge with the res dataframe # Perform the left joins and rename columns res_updated <- res %>% left_join(go_terms, by = "GeneID") %>% left_join(ec_terms, by = "GeneID") %>% dplyr::select(-EC.x, -GOs.x) %>% dplyr::rename(EC = EC.y, GOs = GOs.y) # Filter up-regulated genes up_regulated <- res_updated[res_updated$log2FoldChange > 2 & res_updated$padj < 0.05, ] # Filter down-regulated genes down_regulated <- res_updated[res_updated$log2FoldChange < -2 & res_updated$padj < 0.05, ] # Create a new workbook wb <- createWorkbook() # Add the complete dataset as the first sheet (with annotations) addWorksheet(wb, "Complete") writeData(wb, "Complete_Data", res_updated) # Add the up-regulated genes as the second sheet (with annotations) addWorksheet(wb, "Up_Regulated") writeData(wb, "Up_Regulated", up_regulated) # Add the down-regulated genes as the third sheet (with annotations) addWorksheet(wb, "Down_Regulated") writeData(wb, "Down_Regulated", down_regulated) # Save the workbook to a file #saveWorkbook(wb, "Gene_Expression_with_Annotations_deltasbp_TSB_4h_vs_WT_TSB_4h.xlsx", overwrite = TRUE) #NOTE: The generated annotation-files contains all columns of DESeq2 (GeneName, GeneID, baseMean, log2FoldChange, lfcSE, stat, pvalue, padj) + almost all columns of eggNOG (GeneID, seed_ortholog, evalue, score, eggNOG_OGs, max_annot_lvl, COG_category, Description, Preferred_name, KEGG_ko, KEGG_Pathway, KEGG_Module, KEGG_Reaction, KEGG_rclass, BRITE, KEGG_TC, CAZy, BiGG_Reaction, PFAMs) except for -[GOs, EC] + two columns from Blast2GO (COs, EC); In the code below, we use the columns KEGG_ko and GOs for the KEGG and GO enrichments. #TODO: for Michelle's data, we can also perform both KEGG and GO enrichments. # Set GeneName as row names after the join rownames(res_updated) <- res_updated$GeneName res_updated <- res_updated %>% dplyr::select(-GeneName) ## Set the 'GeneName' column as row.names #rownames(res_updated) <- res_updated$GeneName ## Drop the 'GeneName' column since it's now the row names #res_updated$GeneName <- NULL # -- BREAK_1 -- # ---- Perform KEGG enrichment analysis (up_regulated) ---- gene_list_kegg_up <- up_regulated$KEGG_ko gene_list_kegg_up <- gsub("ko:", "", gene_list_kegg_up) kegg_enrichment_up <- enrichKEGG(gene = gene_list_kegg_up, organism = 'ko') # -- convert the GeneID (Kxxxxxx) to the true GeneID -- # Step 0: Create KEGG to GeneID mapping kegg_to_geneid_up <- up_regulated %>% dplyr::select(KEGG_ko, GeneID) %>% filter(!is.na(KEGG_ko)) %>% # Remove missing KEGG KO entries mutate(KEGG_ko = str_remove(KEGG_ko, "ko:")) # Remove 'ko:' prefix if present # Step 1: Clean KEGG_ko values (separate multiple KEGG IDs) kegg_to_geneid_clean <- kegg_to_geneid_up %>% mutate(KEGG_ko = str_remove_all(KEGG_ko, "ko:")) %>% # Remove 'ko:' prefixes separate_rows(KEGG_ko, sep = ",") %>% # Ensure each KEGG ID is on its own row filter(KEGG_ko != "-") %>% # Remove invalid KEGG IDs ("-") distinct() # Remove any duplicate mappings # Step 2.1: Expand geneID column in kegg_enrichment_up expanded_kegg <- kegg_enrichment_up %>% as.data.frame() %>% separate_rows(geneID, sep = "/") %>% left_join(kegg_to_geneid_clean, by = c("geneID" = "KEGG_ko"), relationship = "many-to-many") %>% # Explicitly handle many-to-many distinct() %>% # Remove duplicate matches group_by(ID) %>% summarise(across(everything(), ~ paste(unique(na.omit(.)), collapse = "/")), .groups = "drop") # Re-collapse results #dplyr::glimpse(expanded_kegg) # Step 3.1: Replace geneID column in the original dataframe kegg_enrichment_up_df <- as.data.frame(kegg_enrichment_up) # Remove old geneID column and merge new one kegg_enrichment_up_df <- kegg_enrichment_up_df %>% dplyr::select(-geneID) %>% left_join(expanded_kegg %>% dplyr::select(ID, GeneID), by = "ID") %>% dplyr::rename(geneID = GeneID) # Rename column back to geneID # ---- Perform KEGG enrichment analysis (down_regulated) ---- # Step 1: Extract KEGG KO terms from down-regulated genes gene_list_kegg_down <- down_regulated$KEGG_ko gene_list_kegg_down <- gsub("ko:", "", gene_list_kegg_down) # Step 2: Perform KEGG enrichment analysis kegg_enrichment_down <- enrichKEGG(gene = gene_list_kegg_down, organism = 'ko') # --- Convert KEGG gene IDs (Kxxxxxx) to actual GeneIDs --- # Step 3: Create KEGG to GeneID mapping from down_regulated dataset kegg_to_geneid_down <- down_regulated %>% dplyr::select(KEGG_ko, GeneID) %>% filter(!is.na(KEGG_ko)) %>% # Remove missing KEGG KO entries mutate(KEGG_ko = str_remove(KEGG_ko, "ko:")) # Remove 'ko:' prefix if present # -- BREAK_2 -- # Step 4: Clean KEGG_ko values (handle multiple KEGG IDs) kegg_to_geneid_down_clean <- kegg_to_geneid_down %>% mutate(KEGG_ko = str_remove_all(KEGG_ko, "ko:")) %>% # Remove 'ko:' prefixes separate_rows(KEGG_ko, sep = ",") %>% # Ensure each KEGG ID is on its own row filter(KEGG_ko != "-") %>% # Remove invalid KEGG IDs ("-") distinct() # Remove duplicate mappings # Step 5: Expand geneID column in kegg_enrichment_down expanded_kegg_down <- kegg_enrichment_down %>% as.data.frame() %>% separate_rows(geneID, sep = "/") %>% # Split multiple KEGG IDs (Kxxxxx) left_join(kegg_to_geneid_down_clean, by = c("geneID" = "KEGG_ko"), relationship = "many-to-many") %>% # Handle many-to-many mappings distinct() %>% # Remove duplicate matches group_by(ID) %>% summarise(across(everything(), ~ paste(unique(na.omit(.)), collapse = "/")), .groups = "drop") # Re-collapse results # Step 6: Replace geneID column in the original kegg_enrichment_down dataframe kegg_enrichment_down_df <- as.data.frame(kegg_enrichment_down) %>% dplyr::select(-geneID) %>% # Remove old geneID column left_join(expanded_kegg_down %>% dplyr::select(ID, GeneID), by = "ID") %>% # Merge new GeneID column dplyr::rename(geneID = GeneID) # Rename column back to geneID # View the updated dataframe head(kegg_enrichment_down_df) # Create a new workbook #wb <- createWorkbook() # Save enrichment results to the workbook addWorksheet(wb, "KEGG_Enrichment_Up") writeData(wb, "KEGG_Enrichment_Up", as.data.frame(kegg_enrichment_up_df)) # Save enrichment results to the workbook addWorksheet(wb, "KEGG_Enrichment_Down") writeData(wb, "KEGG_Enrichment_Down", as.data.frame(kegg_enrichment_down_df)) # Define gene list (up-regulated genes) gene_list_go_up <- up_regulated$GeneID # Extract the 149 up-regulated genes gene_list_go_down <- down_regulated$GeneID # Extract the 65 down-regulated genes # Define background gene set (all genes in res) background_genes <- res_updated$GeneID # Extract the 3646 background genes # Prepare GO annotation data from res go_annotation <- res_updated[, c("GOs","GeneID")] # Extract relevant columns go_annotation <- go_annotation %>% tidyr::separate_rows(GOs, sep = ",") # Split multiple GO terms into separate rows # -- BREAK_3 -- go_enrichment_up <- enricher( gene = gene_list_go_up, # Up-regulated genes TERM2GENE = go_annotation, # Custom GO annotation pvalueCutoff = 0.05, # Significance threshold pAdjustMethod = "BH", universe = background_genes # Define the background gene set ) go_enrichment_up <- as.data.frame(go_enrichment_up) go_enrichment_down <- enricher( gene = gene_list_go_down, # Up-regulated genes TERM2GENE = go_annotation, # Custom GO annotation pvalueCutoff = 0.05, # Significance threshold pAdjustMethod = "BH", universe = background_genes # Define the background gene set ) go_enrichment_down <- as.data.frame(go_enrichment_down) ## Remove the 'p.adjust' column since no adjusted methods have been applied --> In this version we have used pvalue filtering (see above)! #go_enrichment_up <- go_enrichment_up[, !names(go_enrichment_up) %in% "p.adjust"] # Update the Description column with the term descriptions go_enrichment_up$Description <- sapply(go_enrichment_up$ID, function(go_id) { # Using select to get the term description term <- tryCatch({ AnnotationDbi::select(GO.db, keys = go_id, columns = "TERM", keytype = "GOID") }, error = function(e) { message(paste("Error for GO term:", go_id)) # Print which GO ID caused the error return(data.frame(TERM = NA)) # In case of error, return NA }) if (nrow(term) > 0) { return(term$TERM) } else { return(NA) # If no description found, return NA } }) ## Print the updated data frame #print(go_enrichment_up) ## Remove the 'p.adjust' column since no adjusted methods have been applied --> In this version we have used pvalue filtering (see above)! #go_enrichment_down <- go_enrichment_down[, !names(go_enrichment_down) %in% "p.adjust"] # Update the Description column with the term descriptions go_enrichment_down$Description <- sapply(go_enrichment_down$ID, function(go_id) { # Using select to get the term description term <- tryCatch({ AnnotationDbi::select(GO.db, keys = go_id, columns = "TERM", keytype = "GOID") }, error = function(e) { message(paste("Error for GO term:", go_id)) # Print which GO ID caused the error return(data.frame(TERM = NA)) # In case of error, return NA }) if (nrow(term) > 0) { return(term$TERM) } else { return(NA) # If no description found, return NA } }) addWorksheet(wb, "GO_Enrichment_Up") writeData(wb, "GO_Enrichment_Up", as.data.frame(go_enrichment_up)) addWorksheet(wb, "GO_Enrichment_Down") writeData(wb, "GO_Enrichment_Down", as.data.frame(go_enrichment_down)) # Save the workbook with enrichment results saveWorkbook(wb, "DEG_KEGG_GO_deltasbp_TSB_2h_vs_WT_TSB_2h.xlsx", overwrite = TRUE) #Error for GO term: GO:0006807: replace "GO:0006807 obsolete nitrogen compound metabolic process" #obsolete nitrogen compound metabolic process #https://www.ebi.ac.uk/QuickGO/term/GO:0006807 #TODO: marked the color as yellow if the p.adjusted <= 0.05 in GO_enrichment! #mv KEGG_and_GO_Enrichments_Urine_vs_MHB.xlsx KEGG_and_GO_Enrichments_Mac_vs_LB.xlsx #Mac_vs_LB #LB.AB_vs_LB.WT19606 #LB.IJ_vs_LB.WT19606 #LB.W1_vs_LB.WT19606 #LB.Y1_vs_LB.WT19606 #Mac.AB_vs_Mac.WT19606 #Mac.IJ_vs_Mac.WT19606 #Mac.W1_vs_Mac.WT19606 #Mac.Y1_vs_Mac.WT19606 #TODO: write reply hints in KEGG_and_GO_Enrichments_deltasbp_TSB_4h_vs_WT_TSB_4h.xlsx contains icaABCD, gtf1 and gtf2. -
(DEBUG) Draw the Venn diagram to compare the total DEGs across AUM, Urine, and MHB, irrespective of up- or down-regulation.
library(openxlsx) # Function to read and clean gene ID files read_gene_ids <- function(file_path) { # Read the gene IDs from the file gene_ids <- readLines(file_path) # Remove any quotes and trim whitespaces gene_ids <- gsub('"', '', gene_ids) # Remove quotes gene_ids <- trimws(gene_ids) # Trim whitespaces # Remove empty entries or NAs gene_ids <- gene_ids[gene_ids != "" & !is.na(gene_ids)] return(gene_ids) } # Example list of LB files with both -up.id and -down.id for each condition lb_files_up <- c("LB.AB_vs_LB.WT19606-up.id", "LB.IJ_vs_LB.WT19606-up.id", "LB.W1_vs_LB.WT19606-up.id", "LB.Y1_vs_LB.WT19606-up.id") lb_files_down <- c("LB.AB_vs_LB.WT19606-down.id", "LB.IJ_vs_LB.WT19606-down.id", "LB.W1_vs_LB.WT19606-down.id", "LB.Y1_vs_LB.WT19606-down.id") # Combine both up and down files for each condition lb_files <- c(lb_files_up, lb_files_down) # Read gene IDs for each file in LB group #lb_degs <- setNames(lapply(lb_files, read_gene_ids), gsub("-(up|down).id", "", lb_files)) lb_degs <- setNames(lapply(lb_files, read_gene_ids), make.unique(gsub("-(up|down).id", "", lb_files))) lb_degs_ <- list() combined_set <- c(lb_degs[["LB.AB_vs_LB.WT19606"]], lb_degs[["LB.AB_vs_LB.WT19606.1"]]) #unique_combined_set <- unique(combined_set) lb_degs_$AB <- combined_set combined_set <- c(lb_degs[["LB.IJ_vs_LB.WT19606"]], lb_degs[["LB.IJ_vs_LB.WT19606.1"]]) lb_degs_$IJ <- combined_set combined_set <- c(lb_degs[["LB.W1_vs_LB.WT19606"]], lb_degs[["LB.W1_vs_LB.WT19606.1"]]) lb_degs_$W1 <- combined_set combined_set <- c(lb_degs[["LB.Y1_vs_LB.WT19606"]], lb_degs[["LB.Y1_vs_LB.WT19606.1"]]) lb_degs_$Y1 <- combined_set # Example list of Mac files with both -up.id and -down.id for each condition mac_files_up <- c("Mac.AB_vs_Mac.WT19606-up.id", "Mac.IJ_vs_Mac.WT19606-up.id", "Mac.W1_vs_Mac.WT19606-up.id", "Mac.Y1_vs_Mac.WT19606-up.id") mac_files_down <- c("Mac.AB_vs_Mac.WT19606-down.id", "Mac.IJ_vs_Mac.WT19606-down.id", "Mac.W1_vs_Mac.WT19606-down.id", "Mac.Y1_vs_Mac.WT19606-down.id") # Combine both up and down files for each condition in Mac group mac_files <- c(mac_files_up, mac_files_down) # Read gene IDs for each file in Mac group mac_degs <- setNames(lapply(mac_files, read_gene_ids), make.unique(gsub("-(up|down).id", "", mac_files))) mac_degs_ <- list() combined_set <- c(mac_degs[["Mac.AB_vs_Mac.WT19606"]], mac_degs[["Mac.AB_vs_Mac.WT19606.1"]]) mac_degs_$AB <- combined_set combined_set <- c(mac_degs[["Mac.IJ_vs_Mac.WT19606"]], mac_degs[["Mac.IJ_vs_Mac.WT19606.1"]]) mac_degs_$IJ <- combined_set combined_set <- c(mac_degs[["Mac.W1_vs_Mac.WT19606"]], mac_degs[["Mac.W1_vs_Mac.WT19606.1"]]) mac_degs_$W1 <- combined_set combined_set <- c(mac_degs[["Mac.Y1_vs_Mac.WT19606"]], mac_degs[["Mac.Y1_vs_Mac.WT19606.1"]]) mac_degs_$Y1 <- combined_set # Function to clean sheet names to ensure no sheet name exceeds 31 characters truncate_sheet_name <- function(names_list) { sapply(names_list, function(name) { if (nchar(name) > 31) { return(substr(name, 1, 31)) # Truncate sheet name to 31 characters } return(name) }) } # Assuming lb_degs_ is already a list of gene sets (LB.AB, LB.IJ, etc.) # Define intersections between different conditions for LB inter_lb_ab_ij <- intersect(lb_degs_$AB, lb_degs_$IJ) inter_lb_ab_w1 <- intersect(lb_degs_$AB, lb_degs_$W1) inter_lb_ab_y1 <- intersect(lb_degs_$AB, lb_degs_$Y1) inter_lb_ij_w1 <- intersect(lb_degs_$IJ, lb_degs_$W1) inter_lb_ij_y1 <- intersect(lb_degs_$IJ, lb_degs_$Y1) inter_lb_w1_y1 <- intersect(lb_degs_$W1, lb_degs_$Y1) # Define intersections between three conditions for LB inter_lb_ab_ij_w1 <- Reduce(intersect, list(lb_degs_$AB, lb_degs_$IJ, lb_degs_$W1)) inter_lb_ab_ij_y1 <- Reduce(intersect, list(lb_degs_$AB, lb_degs_$IJ, lb_degs_$Y1)) inter_lb_ab_w1_y1 <- Reduce(intersect, list(lb_degs_$AB, lb_degs_$W1, lb_degs_$Y1)) inter_lb_ij_w1_y1 <- Reduce(intersect, list(lb_degs_$IJ, lb_degs_$W1, lb_degs_$Y1)) # Define intersection between all four conditions for LB inter_lb_ab_ij_w1_y1 <- Reduce(intersect, list(lb_degs_$AB, lb_degs_$IJ, lb_degs_$W1, lb_degs_$Y1)) # Now remove the intersected genes from each original set for LB venn_list_lb <- list() # For LB.AB, remove genes that are also in other conditions venn_list_lb[["LB.AB_only"]] <- setdiff(lb_degs_$AB, union(inter_lb_ab_ij, union(inter_lb_ab_w1, inter_lb_ab_y1))) # For LB.IJ, remove genes that are also in other conditions venn_list_lb[["LB.IJ_only"]] <- setdiff(lb_degs_$IJ, union(inter_lb_ab_ij, union(inter_lb_ij_w1, inter_lb_ij_y1))) # For LB.W1, remove genes that are also in other conditions venn_list_lb[["LB.W1_only"]] <- setdiff(lb_degs_$W1, union(inter_lb_ab_w1, union(inter_lb_ij_w1, inter_lb_ab_w1_y1))) # For LB.Y1, remove genes that are also in other conditions venn_list_lb[["LB.Y1_only"]] <- setdiff(lb_degs_$Y1, union(inter_lb_ab_y1, union(inter_lb_ij_y1, inter_lb_ab_w1_y1))) # Add the intersections for LB (same as before) venn_list_lb[["LB.AB_AND_LB.IJ"]] <- inter_lb_ab_ij venn_list_lb[["LB.AB_AND_LB.W1"]] <- inter_lb_ab_w1 venn_list_lb[["LB.AB_AND_LB.Y1"]] <- inter_lb_ab_y1 venn_list_lb[["LB.IJ_AND_LB.W1"]] <- inter_lb_ij_w1 venn_list_lb[["LB.IJ_AND_LB.Y1"]] <- inter_lb_ij_y1 venn_list_lb[["LB.W1_AND_LB.Y1"]] <- inter_lb_w1_y1 # Define intersections between three conditions for LB venn_list_lb[["LB.AB_AND_LB.IJ_AND_LB.W1"]] <- inter_lb_ab_ij_w1 venn_list_lb[["LB.AB_AND_LB.IJ_AND_LB.Y1"]] <- inter_lb_ab_ij_y1 venn_list_lb[["LB.AB_AND_LB.W1_AND_LB.Y1"]] <- inter_lb_ab_w1_y1 venn_list_lb[["LB.IJ_AND_LB.W1_AND_LB.Y1"]] <- inter_lb_ij_w1_y1 # Define intersection between all four conditions for LB venn_list_lb[["LB.AB_AND_LB.IJ_AND_LB.W1_AND_LB.Y1"]] <- inter_lb_ab_ij_w1_y1 # Assuming mac_degs_ is already a list of gene sets (Mac.AB, Mac.IJ, etc.) # Define intersections between different conditions inter_mac_ab_ij <- intersect(mac_degs_$AB, mac_degs_$IJ) inter_mac_ab_w1 <- intersect(mac_degs_$AB, mac_degs_$W1) inter_mac_ab_y1 <- intersect(mac_degs_$AB, mac_degs_$Y1) inter_mac_ij_w1 <- intersect(mac_degs_$IJ, mac_degs_$W1) inter_mac_ij_y1 <- intersect(mac_degs_$IJ, mac_degs_$Y1) inter_mac_w1_y1 <- intersect(mac_degs_$W1, mac_degs_$Y1) # Define intersections between three conditions inter_mac_ab_ij_w1 <- Reduce(intersect, list(mac_degs_$AB, mac_degs_$IJ, mac_degs_$W1)) inter_mac_ab_ij_y1 <- Reduce(intersect, list(mac_degs_$AB, mac_degs_$IJ, mac_degs_$Y1)) inter_mac_ab_w1_y1 <- Reduce(intersect, list(mac_degs_$AB, mac_degs_$W1, mac_degs_$Y1)) inter_mac_ij_w1_y1 <- Reduce(intersect, list(mac_degs_$IJ, mac_degs_$W1, mac_degs_$Y1)) # Define intersection between all four conditions inter_mac_ab_ij_w1_y1 <- Reduce(intersect, list(mac_degs_$AB, mac_degs_$IJ, mac_degs_$W1, mac_degs_$Y1)) # Now remove the intersected genes from each original set venn_list_mac <- list() # For Mac.AB, remove genes that are also in other conditions venn_list_mac[["Mac.AB_only"]] <- setdiff(mac_degs_$AB, union(inter_mac_ab_ij, union(inter_mac_ab_w1, inter_mac_ab_y1))) # For Mac.IJ, remove genes that are also in other conditions venn_list_mac[["Mac.IJ_only"]] <- setdiff(mac_degs_$IJ, union(inter_mac_ab_ij, union(inter_mac_ij_w1, inter_mac_ij_y1))) # For Mac.W1, remove genes that are also in other conditions venn_list_mac[["Mac.W1_only"]] <- setdiff(mac_degs_$W1, union(inter_mac_ab_w1, union(inter_mac_ij_w1, inter_mac_ab_w1_y1))) # For Mac.Y1, remove genes that are also in other conditions venn_list_mac[["Mac.Y1_only"]] <- setdiff(mac_degs_$Y1, union(inter_mac_ab_y1, union(inter_mac_ij_y1, inter_mac_ab_w1_y1))) # Add the intersections (same as before) venn_list_mac[["Mac.AB_AND_Mac.IJ"]] <- inter_mac_ab_ij venn_list_mac[["Mac.AB_AND_Mac.W1"]] <- inter_mac_ab_w1 venn_list_mac[["Mac.AB_AND_Mac.Y1"]] <- inter_mac_ab_y1 venn_list_mac[["Mac.IJ_AND_Mac.W1"]] <- inter_mac_ij_w1 venn_list_mac[["Mac.IJ_AND_Mac.Y1"]] <- inter_mac_ij_y1 venn_list_mac[["Mac.W1_AND_Mac.Y1"]] <- inter_mac_w1_y1 # Define intersections between three conditions venn_list_mac[["Mac.AB_AND_Mac.IJ_AND_Mac.W1"]] <- inter_mac_ab_ij_w1 venn_list_mac[["Mac.AB_AND_Mac.IJ_AND_Mac.Y1"]] <- inter_mac_ab_ij_y1 venn_list_mac[["Mac.AB_AND_Mac.W1_AND_Mac.Y1"]] <- inter_mac_ab_w1_y1 venn_list_mac[["Mac.IJ_AND_Mac.W1_AND_Mac.Y1"]] <- inter_mac_ij_w1_y1 # Define intersection between all four conditions venn_list_mac[["Mac.AB_AND_Mac.IJ_AND_Mac.W1_AND_Mac.Y1"]] <- inter_mac_ab_ij_w1_y1 # Save the gene IDs to Excel for further inspection (optional) write.xlsx(lb_degs, file = "LB_DEGs.xlsx") write.xlsx(mac_degs, file = "Mac_DEGs.xlsx") # Clean sheet names and write the Venn intersection sets for LB and Mac groups into Excel files write.xlsx(venn_list_lb, file = "Venn_LB_Genes_Intersect.xlsx", sheetName = truncate_sheet_name(names(venn_list_lb)), rowNames = FALSE) write.xlsx(venn_list_mac, file = "Venn_Mac_Genes_Intersect.xlsx", sheetName = truncate_sheet_name(names(venn_list_mac)), rowNames = FALSE) # Venn Diagram for LB group venn1 <- ggvenn(lb_degs_, fill_color = c("skyblue", "tomato", "gold", "orchid"), stroke_size = 0.4, set_name_size = 5) ggsave("Venn_LB_Genes.png", plot = venn1, width = 7, height = 7, dpi = 300) # Venn Diagram for Mac group venn2 <- ggvenn(mac_degs_, fill_color = c("lightgreen", "slateblue", "plum", "orange"), stroke_size = 0.4, set_name_size = 5) ggsave("Venn_Mac_Genes.png", plot = venn2, width = 7, height = 7, dpi = 300) cat("✅ All Venn intersection sets exported to Excel successfully.\n")