/media/jhuang/INTENSO
(empty; data now on ~/DATA_Intenso)
~/DATA
| # |
Name |
| 1 |
Data_Ute_MKL1 |
| 2 |
Data_Ute_RNA_4_2022-11_test |
| 3 |
Data_Ute_RNA_3 |
| 4 |
Data_Susanne_Carotis_RNASeq_PUBLISHING |
| 5 |
Data_Jiline_Yersinia_SNP |
| 6 |
Data_Tam_ABAYE_RS05070_on_A_calcoaceticus_baumannii_complex_DUPLICATED_DEL |
| 7 |
Data_Nicole_CRC1648 |
| 8 |
Mouse_HS3ST1_12373_out |
| 9 |
Mouse_HS3ST1_12175_out |
| 10 |
Data_Biobakery |
| 11 |
Data_Xiaobo_10x_2 |
| 12 |
Data_Xiaobo_10x_3 |
| 13 |
Talk_Nicole_CRC1648 |
| 14 |
Talks_Bioinformatics_Meeting |
| 15 |
Talks_resources |
| 16 |
Data_Susanne_MPox_DAMIAN |
| 17 |
Data_host_transcriptional_response |
| 18 |
Talks_including_DEEP-DV |
| 19 |
DOKTORARBEIT |
| 20 |
Data_Susanne_MPox |
| 21 |
Data_Jiline_Transposon |
| 22 |
Data_Jiline_Transposon2 |
| 23 |
Data_Matlab |
| 24 |
deepseek-ai |
| 25 |
Stick_Mi_DEL |
| 26 |
TODO_shares |
| 27 |
Data_Ute_RNA_4 |
| 28 |
Data_Liu_PCA_plot |
| 29 |
README_run_viral-ngs_inside_Docker |
| 30 |
README_compare_genomes |
| 31 |
mapped.bam |
| 32 |
Data_Serpapi |
| 33 |
Data_Ute_RNA_1_2 |
| 34 |
Data_Marc_RNAseq_2024 |
| 35 |
Data_Nicole_CaptureProbeSequencing |
| 36 |
LOG_mapping |
| 37 |
Data_Huang_Human_herpesvirus_3 |
| 38 |
Data_Nicole_DAMIAN_Post-processing_Pathoprobe_FluB_Links |
| 39 |
Access_to_Win7 |
| 40 |
Data_DAMIAN_Post-processing_Flavivirus_and_FSME_and_Haemophilus |
| 41 |
Data_Luise_Sepi_STKN |
| 42 |
Data_Patricia_Sepi_7samples |
| 43 |
Data_Soeren_2025_PUBLISHING |
| 44 |
Data_Ben_RNAseq_2025 |
| 45 |
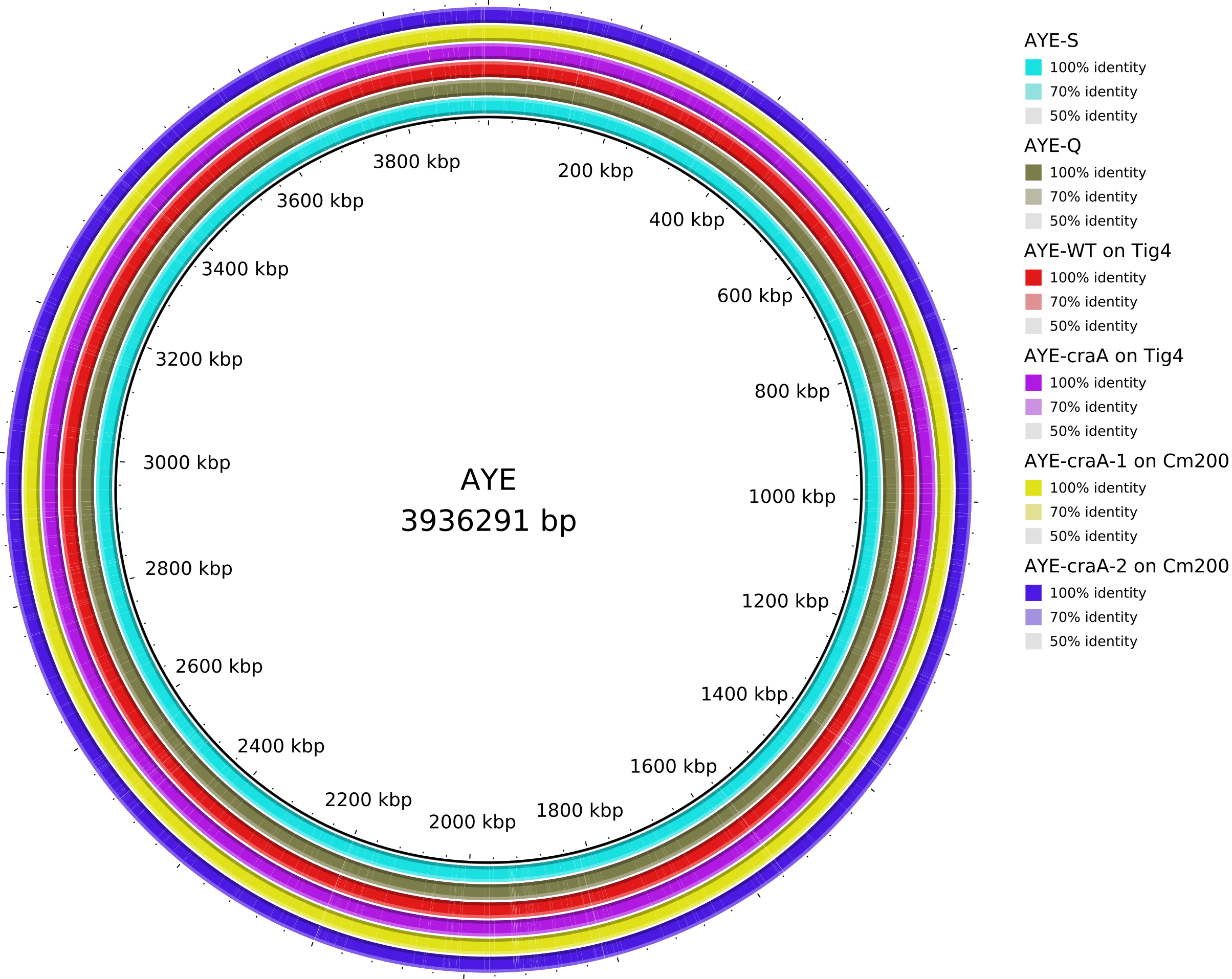
Data_Tam_DNAseq_2025_AYE-WT_Q_S_craA-Tig4_craA-1-Cm200_craA-2-Cm200 |
| 46 |
Data_Patricia_Transposon |
| 47 |
Data_Patricia_Transposon_2025 |
| 48 |
Colocation_Space |
| 49 |
Data_Tam_Methylation_2025_empty |
| 50 |
2025-11-03_eVB-Schreiben_12-57.pdf |
| 51 |
DEGs_Group1_A1-A3+A8-A10_vs_Group2_B10-B16.png |
| 52 |
README.pdf |
| 53 |
Data_Hannes_JCM00612 |
| 54 |
167_redundant_DEL |
| 55 |
Lehre_Bioinformatik |
| 56 |
Data_Ben_Boruta_Analysis |
| 57 |
Data_Childrensclinic_16S_2025_DEL |
| 58 |
Data_Ben_Mycobacterium_pseudoscrofulaceum |
| 59 |
Foong_RNA_mSystems_Huang_Changed.txt |
| 60 |
Data_Pietro_Scatturo_and_Charlotte_Uetrecht_16S_2025 |
| 61 |
Data_JuliaBerger_RNASeq_SARS-CoV-2 |
| 62 |
Data_PaulBongarts_S.epidermidis_HDRNA |
| 63 |
Data_Ute |
| 64 |
Data_Foong_DNAseq_2025_AYE_Dark_vs_Light_TODO |
| 65 |
Data_Foong_RNAseq_2021_ATCC19606_Cm |
| 66 |
Data_Tam_Funding |
| 67 |
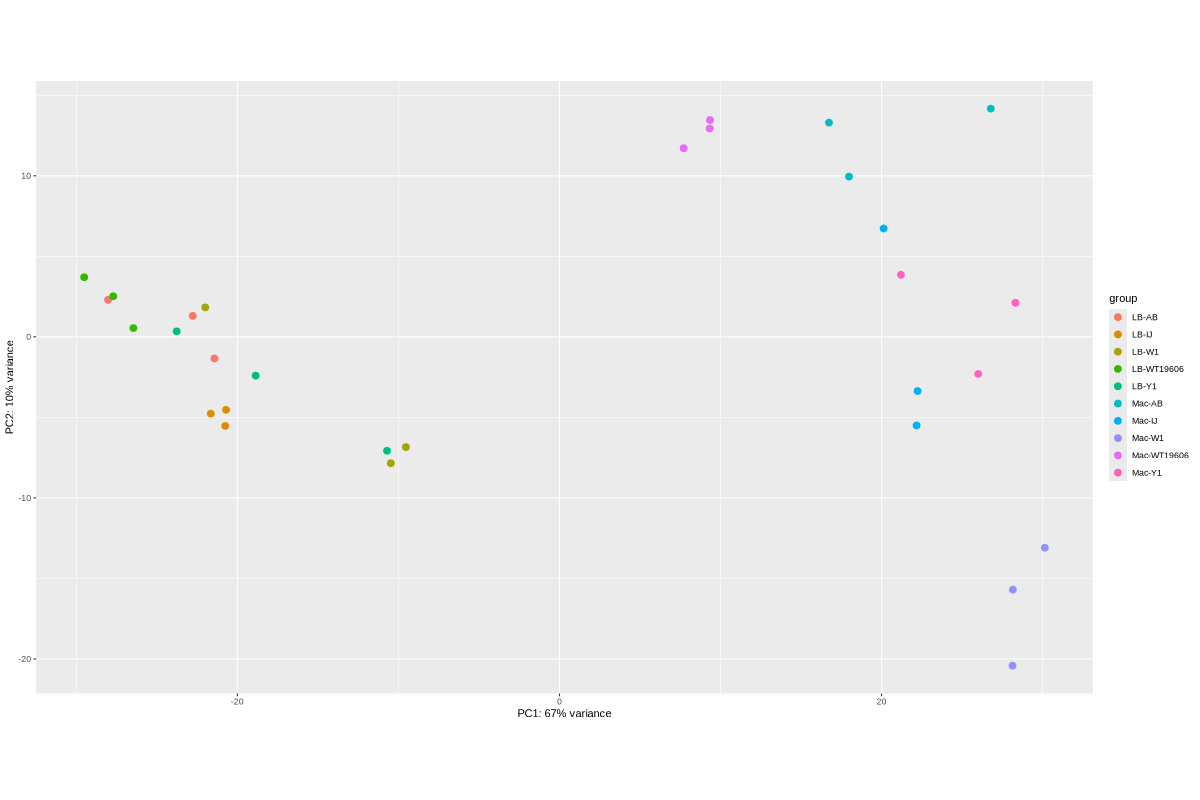
Data_Tam_RNAseq_2025_LB-AB_IJ_W1_Y1_WT_vs_Mac-AB_IJ_W1_Y1_WT_on_ATCC19606 |
| 68 |
Data_Tam_RNAseq_2025_subMIC_exposure_on_ATCC19606 |
| 69 |
Data_Tam.txt |
| 70 |
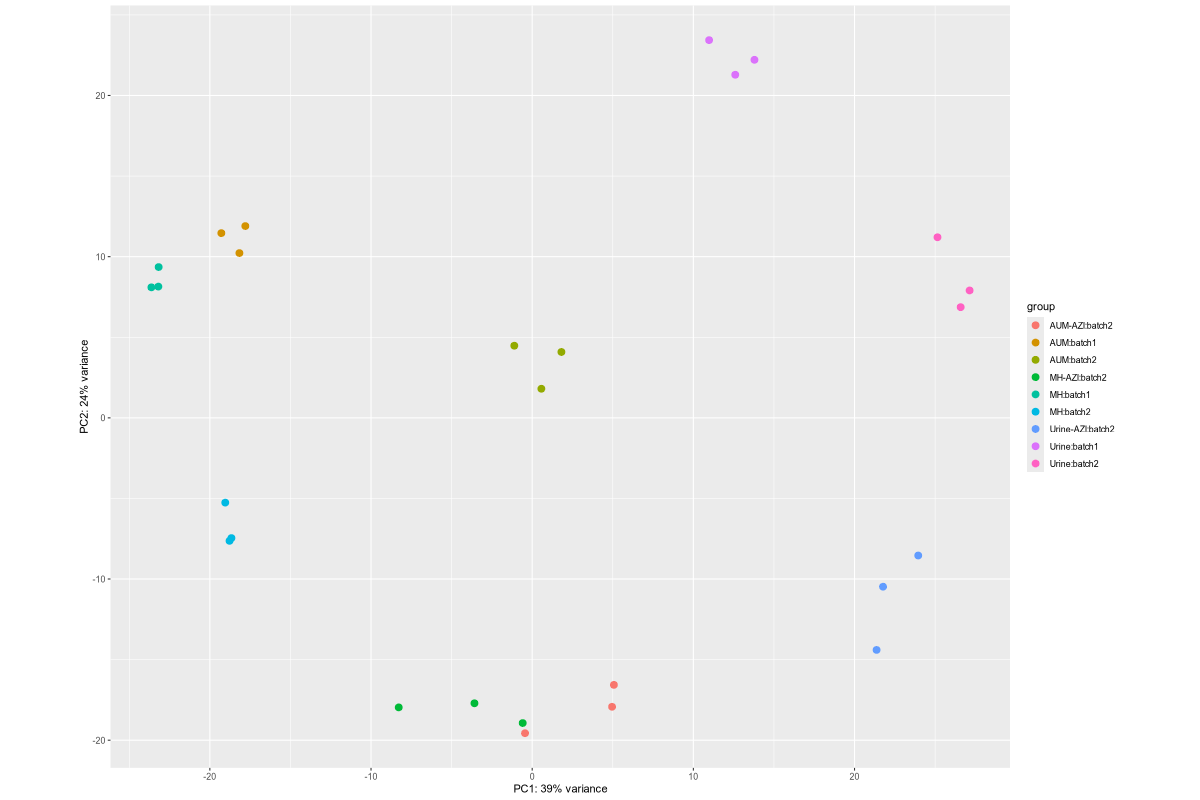
Data_Tam_RNAseq_2024_AUM_MHB_Urine_on_ATCC19606 |
| 71 |
Data_Tam_Metagenomics_2026 |
| 72 |
Data_Michelle |
| 73 |
Data_Nicole_16S_2025_Childrensclinic |
| 74 |
Data_Sophie_HDV_Sequences |
| 75 |
Data_Tam_DNAseq_2026_19606deltaIJfluE |
| 76 |
README_nf-core |
| 77 |
Data_Vero_Kymographs |
| 78 |
Access_to_Win10 |
| 79 |
Data_Patricia_AMRFinderPlus_2025 |
| 80 |
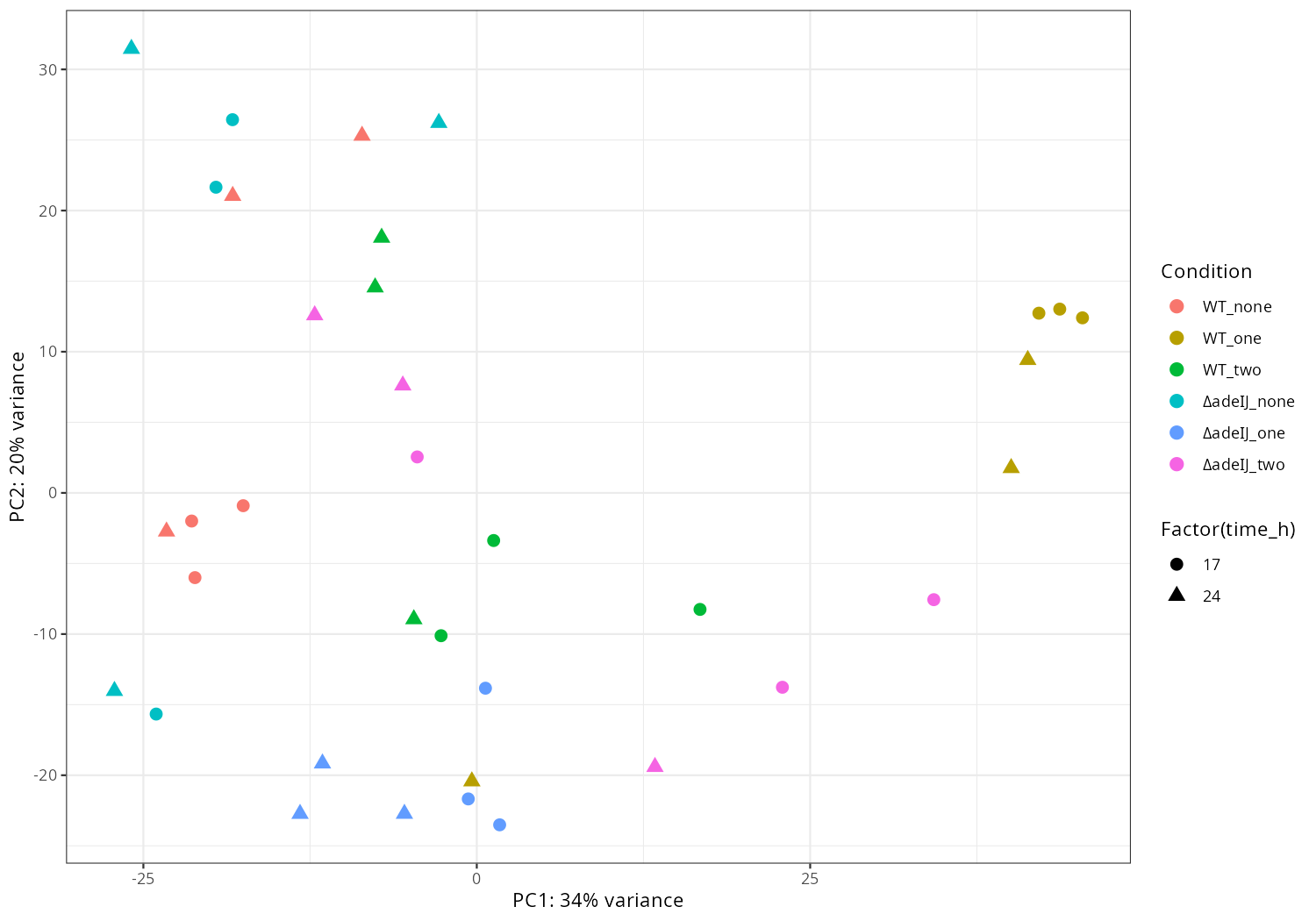
Data_Tam_DNAseq_2025_Unknown-adeABadeIJ_adeIJK_CM1_CM2 |
| 81 |
Data_Damian |
| 82 |
Data_Karoline_16S |
| 83 |
Data_JuliaFuchs_RNAseq_2025 |
| 84 |
Data_Tam_DNAseq_2025_ATCC19606-Y1Y2Y3Y4W1W2W3W4_TODO |
| 85 |
Data_Tam_DNAseq_2026_Acinetobacter_harbinensis |
| 86 |
Data_Benjamin_DNAseq_2026_GE11174 |
| 87 |
Data_Susanne_spatialRNA_2022.9.1_backup |
| 88 |
Data_Susanne_spatialRNA |
~/DATA_A
| # |
Name |
| 1 |
Data_Damian_NEW_CREATED |
| 2 |
Data_R_bubbleplots |
| 3 |
Data_Ute_TRANSFERED_DEL |
| 4 |
Paper_Target_capture_sequencing_MHH_PUBLISHED |
| 5 |
Data_Nicole8_Lamprecht_new_PUBLISHED |
| 6 |
Data_Samira_RNAseq |
~/DATA_B
| # |
Name |
| 1 |
Data_DAMIAN_endocarditis_encephalitis |
| 2 |
Data_Denise_sT_PUBLISHING |
| 3 |
Data_Fran2_16S_func |
| 4 |
Data_Holger_5179-R1_vs_5179 |
| 5 |
Antraege_ |
| 6 |
Data_16S_Nicole_210222 |
| 7 |
Data_Adam_Influenza_A_virus |
| 8 |
Data_Anna_Efaecium_assembly |
| 9 |
Data_Bactopia |
| 10 |
Data_Ben_RNAseq |
| 11 |
Data_Johannes_PIV3 |
| 12 |
Data_Luise_Epidome_longitudinal_nose |
| 13 |
Data_Manja_Hannes_Probedesign |
| 14 |
Data_Marc_AD_PUBLISHING |
| 15 |
Data_Marc_RNA-seq_Saureus_Review |
| 16 |
Data_Nicole_16S |
| 17 |
Data_Nicole_cfDNA_pathogens |
| 18 |
Data_Ring_and_CSF_PegivirusC_DAMIAN |
| 19 |
Data_Song_Microarray |
| 20 |
Data_Susanne_Omnikron |
| 21 |
Data_Viro |
| 22 |
Doktorarbeit |
| 23 |
Poster_Rohde_20230724 |
| 24 |
Data_Django |
| 25 |
Data_Holger_S.epidermidis_1585_5179_HD05 |
| 26 |
Data_Manja_RNAseq_Organoids_Virus |
| 27 |
Data_Holger_MT880870_MT880872_Annotation |
| 28 |
Data_Soeren_RNA-seq_2022 |
| 29 |
Data_Manja_RNAseq_Organoids_Merged |
| 30 |
Data_Gunnar_Yersiniomics |
| 31 |
Data_Manja_RNAseq_Organoids |
| 32 |
Data_Susanne_Carotis_MS |
~/DATA_C
(names only; as listed)
| # |
Name |
| 1 |
2022-10-27_IRI_manuscript_v03_JH.docx |
| 2 |
16304905.fasta |
| 3 |
’16S data manuscript_NF.docx’ |
| 4 |
180820_2_supp_4265595_sw6zjk.docx |
| 5 |
180820_2_supp_4265596_sw6zjk.docx |
| 6 |
1a_vs_3.csv |
| 7 |
‘2.05.01.05-A01 Urlaubsantrag-Shuting-beantragt.pdf’ |
| 8 |
2014SawickaBBA.pdf |
| 9 |
20160509Manuscript_NDM_OXA_mitKomm.doc |
| 10 |
220607_Agenda_monthly_meeting.pdf |
| 11 |
‘20221129 Table mutations.docx’ |
| 12 |
230602_NB501882_0428_AHKG53BGXT.zip |
| 13 |
362383173.rar |
| 14 |
562.9459.1.fa |
| 15 |
562.9459.1_rc.fa |
| 16 |
ASA3P.pdf |
| 17 |
All_indels_annotated_vHR.xlsx |
| 18 |
‘Amplikon_indeces_Susanne +groups.xlsx’ |
| 19 |
Amplikon_indeces_Susanne.xlsx |
| 20 |
GAMOLA2 |
| 21 |
Data_Susanne_Carotis_spatialRNA_PUBLISHING (dead link) |
| 22 |
Data_Paul_Staphylococcus_epidermidis |
| 23 |
Data_Nicola_Schaltenberg_PICRUSt |
| 24 |
Data_Nicola_Schaltenberg |
| 25 |
Data_Nicola_Gagliani |
| 26 |
Data_methylome_MMc |
| 27 |
Data_Jingang |
| 28 |
Data_Indra_RNASeq_GSM2262901 |
| 29 |
Data_Holger_VRE |
| 30 |
Data_Holger_Pseudomonas_aeruginosa_SNP |
| 31 |
Data_Hannes_ChIPSeq |
| 32 |
Data_Emilia_MeDIP |
| 33 |
Data_ChristophFR_HepE_published |
| 34 |
Data_Christopher_MeDIP_MMc_published |
| 35 |
Data_Anna_Kieler_Sepi_Staemme |
| 36 |
Data_Anna12_HAPDICS_final |
| 37 |
Data_Anastasia_RNASeq_PUBLISHING |
| 38 |
Aufnahmeantrag_komplett_10_2022.pdf |
| 39 |
Astrovirus.pdf |
| 40 |
COMMANDS |
| 41 |
Bacterial_pipelines.txt |
| 42 |
COMPSRA_uke_DEL.jar |
| 43 |
ChIPSeq_pipeline_desc.docx |
| 44 |
ChIPSeq_pipeline_desc.pdf |
| 45 |
Comparative_genomic_analysis_of_eight_novel_haloal.pdf |
| 46 |
CvO_Klassenliste_7_3.pdf |
| 47 |
‘Copy of pool_b1_CGATGT_300.xlsx’ |
| 48 |
Fran_16S_Exp8-17-21-27.txt |
| 49 |
HPI_DRIVE |
| 50 |
HEV_aligned.fasta |
| 51 |
INTENSO_DIR |
| 52 |
HPI_samples_for_NGS_29.09.22.xlsx |
| 53 |
Hotmail_to_Gmail |
| 54 |
Indra_Thesis_161020.pdf |
| 55 |
‘LT K331A.gbk’ |
| 56 |
LOG_p954_stat |
| 57 |
LOG |
| 58 |
Manuscript_10_02_2021.docx |
| 59 |
Metagenomics_Tools_and_Insights.pdf |
| 60 |
‘Miseq Amplikon LAuf April.xlsx’ |
| 61 |
NGS.tar.gz |
| 62 |
Nachweis_Bakterien_Viren_im_Hochdurchsatz.pdf |
| 63 |
Nicole8_Lamprecht_logs |
| 64 |
Nanopore.handouts.pdf |
| 65 |
‘Norovirus paper Susanne 191105.docx’ |
| 66 |
PhyloRNAalifold.pdf |
| 67 |
README_R |
| 68 |
README_RNAHiSwitch_DEL |
| 69 |
RNA-NGS_Analysis_modul3_NanoStringNorm.zip |
| 70 |
RNAConSLOptV1.2.tar.gz |
| 71 |
‘RSV GFP5 including 3`UTR.docx’ |
| 72 |
SNPs_on_pangenome.txt |
| 73 |
SERVER |
| 74 |
R_tutorials-master.zip |
| 75 |
Rawdata_Readme.pdf |
| 76 |
SUB10826945_record_preview.txt |
| 77 |
S_staphylococcus_annotated_diff_expr.xls |
| 78 |
Snakefile_list |
| 79 |
Source_Classification_Code.rds |
| 80 |
Supplementary_Table_S3.xlsx |
| 81 |
Untitled.ipynb |
| 82 |
UniproUGENE_UserManual.pdf |
| 83 |
Untitled1.ipynb |
| 84 |
Untitled2.ipynb |
| 85 |
Untitled3.ipynb |
| 86 |
WAC6h_vs_WAP6h_down.txt |
| 87 |
damian_nodbs |
| 88 |
WAC6h_vs_WAP6h_up.txt |
| 89 |
‘add. Figures Hamburg_UKE.pptx’ |
| 90 |
all_gene_counts_with_annotation.xlsx |
| 91 |
app_flask.py |
| 92 |
bengal-bay-0.1.json |
| 93 |
bengal3_ac3.yml |
| 94 |
call_shell_from_Ruby.png |
| 95 |
bengal3ac3.yml |
| 96 |
empty.fasta |
| 97 |
coefficients_csaw_vs_diffreps.xlsx |
| 98 |
exchange.txt |
| 99 |
exdata-data-NEI_data.zip |
| 100 |
genes_wac6_wap6.xls |
| 101 |
go1.13.linux-amd64.tar.gz.1 |
| 102 |
hev_p2-p5.fa |
| 103 |
map_corrected_backup.txt |
| 104 |
install_nginx_on_hamm |
| 105 |
hg19.rmsk.bed |
| 106 |
metadata-9563675-processed-ok.tsv |
| 107 |
mkg_sprechstundenflyer_ver1b_dezember_2019.pdf |
| 108 |
multiqc_config.yaml |
| 109 |
p11326_OMIKRON3398_corsurv.gb |
| 110 |
p11326_OMIKRON3398_corsurv.gb_converted.fna |
| 111 |
parseGenbank_reformat.py |
| 112 |
pangenome-snakemake-master.zip |
| 113 |
‘phylo tree draft.pdf’ |
| 114 |
qiime_params.txt |
| 115 |
pool_b1_CGATGT_300.zip |
| 116 |
qiime_params_backup.txt |
| 117 |
qiime_params_s16_s18.txt |
| 118 |
snakePipes |
| 119 |
results_description.html |
| 120 |
rnaalihishapes.tar.gz |
| 121 |
rnaseq_length_bias.pdf |
| 122 |
3932-Leber |
| 123 |
BioPython |
| 124 |
Biopython |
| 125 |
DEEP-DV |
| 126 |
DOKTORARBEIT |
| 127 |
Data_16S_Arck_vaginal_stool |
| 128 |
Data_16S_BS052 |
| 129 |
Data_16S_Birgit |
| 130 |
Data_16S_Christner |
| 131 |
Data_16S_Leonie |
| 132 |
Data_16S_PatientA-G_CSF |
| 133 |
Data_16S_Schaltenberg |
| 134 |
Data_16S_benchmark |
| 135 |
Data_16S_benchmark2 |
| 136 |
Data_16S_gcdh_BKV |
| 137 |
Data_Alex1_Amplicon |
| 138 |
Data_Alex1_SNP |
| 139 |
Data_Analysis_for_Life_Science |
| 140 |
Data_Anna13_vanA-Element |
| 141 |
Data_Anna14_PACBIO_methylation |
| 142 |
Data_Anna_C.acnes2_old_DEL |
| 143 |
Data_Anna_MT880872_update |
| 144 |
Data_Anna_gap_filling_agrC |
| 145 |
Data_Baechlein_Hepacivirus_2018 |
| 146 |
Data_Bornavirus |
| 147 |
Data_CSF |
| 148 |
Data_Christine_cz19-178-rothirsch-bovines-hepacivirus |
| 149 |
Data_Daniela_adenovirus_WGS |
| 150 |
Data_Emilia_MeDIP_DEL |
| 151 |
Data_Francesco2021_16S |
| 152 |
Data_Francesco2021_16S_re |
| 153 |
Data_Gunnar_MS |
| 154 |
Data_Hannes_RNASeq |
| 155 |
Data_Holger_Efaecium_variants_PUBLISHED |
| 156 |
Data_Holger_VRE_DEL |
| 157 |
Data_Icebear_Damian |
| 158 |
Data_Indra3_H3K4_p2_DEL |
| 159 |
Data_Indra6_RNASeq_ChipSeq_Integration_DEL |
| 160 |
Data_Indra_Figures |
| 161 |
Data_KatjaGiersch_new_HDV |
| 162 |
Data_MHH_Encephalitits_DAMIAN |
| 163 |
Data_Manja_RPAChIPSeq_public |
| 164 |
Data_Manuel_WGS_Yersinia |
| 165 |
Data_Manuel_WGS_Yersinia2_DEL |
| 166 |
Data_Manuel_WGS_Yersinia_DEL |
| 167 |
Data_Marcus_tracrRNA_structures |
| 168 |
Data_Mausmaki_Damian |
| 169 |
Data_Nicole1_Tropheryma_whipplei |
| 170 |
Data_Nicole5 |
| 171 |
Data_Nicole5_77-92 |
| 172 |
Data_PaulBecher_Rotavirus |
| 173 |
Data_Pietschmann_HCV_Amplicon_bigFile |
| 174 |
Data_Piscine_Orthoreovirus_3_in_Brown_Trout |
| 175 |
Data_Proteomics |
| 176 |
Data_RNABioinformatics |
| 177 |
Data_RNAKinetics |
| 178 |
Data_R_courses |
| 179 |
Data_SARS-CoV-2 |
| 180 |
Data_SARS-CoV-2_Genome_Announcement_PUBLISHED |
| 181 |
Data_Seite |
| 182 |
Data_Song_aggregate_sum |
| 183 |
Data_Susanne_Amplicon_RdRp_orf1_2_re |
| 184 |
Data_Tabea_RNASeq |
| 185 |
Data_Thaiss1_Microarray_new |
| 186 |
Data_Tintelnot_16S |
| 187 |
Data_Wuenee_Plots |
| 188 |
Data_Yang_Poster |
| 189 |
Data_jupnote |
| 190 |
Data_parainfluenza |
| 191 |
Data_snakemake_recipe |
| 192 |
Data_temp |
| 193 |
Data_viGEN |
| 194 |
Genomic_Data_Science |
| 195 |
Learn_UGENE |
| 196 |
MMcPaper |
| 197 |
Manuscript_Epigenetics_Macrophage_Yersinia |
| 198 |
Manuscript_RNAHiSwitch |
| 199 |
MeDIP_Emilia_copy_DEL |
| 200 |
Method_biopython |
| 201 |
NGS |
| 202 |
Okazaki-Seq_Processing |
| 203 |
RNA-NGS_Analysis_modul3_NanoStringNorm |
| 204 |
RNAConSLOptV1.2 |
| 205 |
RNAHeliCes |
| 206 |
RNA_li_HeliCes |
| 207 |
RNAliHeliCes |
| 208 |
RNAliHeliCes_Relatedshapes_modified |
| 209 |
R_refcard |
| 210 |
R_DataCamp |
| 211 |
R_cats_package |
| 212 |
R_tutorials-master |
| 213 |
SnakeChunks |
| 214 |
align_4l_on_FJ705359 |
| 215 |
align_4p_on_FJ705359 |
| 216 |
assembly |
| 217 |
bacto |
| 218 |
bam2fastq_mapping_again |
| 219 |
chipster |
| 220 |
damian_GUI |
| 221 |
enhancer-snakemake-demo |
| 222 |
hg19_gene_annotations |
| 223 |
interlab_comparison_DEL |
| 224 |
my_flask |
| 225 |
papers |
| 226 |
pangenome-snakemake_zhaoc1 |
| 227 |
pyflow-epilogos |
| 228 |
raw_data_rnaseq_Indra |
| 229 |
test_raw_data_dnaseq |
| 230 |
test_raw_data_rnaseq |
| 231 |
to_Francesco |
| 232 |
ukepipe |
| 233 |
ukepipe_nf |
| 234 |
var_www_DjangoApp_mysite2_2023-05 |
| 235 |
roentgenpass.pdf |
| 236 |
salmon_tx2gene_GRCh38.tsv |
| 237 |
salmon_tx2gene_chrHsv1.tsv |
| 238 |
‘sample IDs_Lamprecht.xlsx’ |
| 239 |
summarySCC_PM25.rds |
| 240 |
untitled.py |
| 241 |
tutorial-rnaseq.pdf |
| 242 |
x.log |
| 243 |
webapp.tar.gz |
| 244 |
temp |
| 245 |
temp2 |
| 246 |
Data_Susanne_Amplicon_haplotype_analyses_RdRp_orf1_2_re |
| 247 |
Data_Susanne_WGS_unbiased |
~/DATA_D
| # |
Name |
| 1 |
Data_Soeren_RNA-seq_2023_PUBLISHING |
| 2 |
Data_Ute |
| 3 |
Data_Marc_RNA-seq_Sepidermidis |
| 4 |
Data_Patricia_Transposon |
| 5 |
Books_DA_for_Life |
| 6 |
Data_Sven |
| 7 |
Datasize_calculation_based_on_coverage.txt |
| 8 |
Data_Paul_HD46_1-wt_resequencing |
| 9 |
Data_Sanam_DAMIAN |
| 10 |
Data_Tam_variant_calling |
| 11 |
Data_Samira_Manuscripts |
| 12 |
Data_Silvia_VoltRon_Debug |
| 13 |
Data_Pietschmann_229ECoronavirus_Mutations_2024 |
| 14 |
Data_Pietschmann_229ECoronavirus_Mutations_2025 |
| 15 |
Data_Birthe_Svenja_RSV_Probe3_PUBLISHING |
~/DATA_E
| # |
Name |
| 1 |
j_huang_until_201904 |
| 2 |
Data_2019_April |
| 3 |
Data_2019_May |
| 4 |
Data_2019_June |
| 5 |
Data_2019_July |
| 6 |
Data_2019_August |
| 7 |
Data_2019_September |
| 8 |
Data_Song_RNASeq_PUBLISHED |
| 9 |
Data_Laura_MP_RNASeq |
| 10 |
Data_Nicole6_HEV_Swantje2 |
| 11 |
Data_Becher_Damian_Picornavirus_BovHepV |
| 12 |
bacteria_refseq.zip |
| 13 |
bacteria_refseq |
| 14 |
Data_Rotavirus |
| 15 |
Data_Xiaobo_10x |
| 16 |
Data_Becher_Damian_Picornavirus_BovHepV_INCOMPLETE_DEL |
~/DATA_Intenso
| # |
Name |
| 1 |
HOME_FREIBURG_DEL |
| 2 |
150810_M03701_0019_000000000-AFJFK |
| 3 |
Data_Thaiss2_Microarray |
| 4 |
VirtualBox_VMs_DEL |
| 5 |
‘VirtualBox VMs_DEL’ |
| 6 |
‘VirtualBox VMs2_DEL’ |
| 7 |
websites |
| 8 |
DATA |
| 9 |
Data_Laura |
| 10 |
Data_Laura_2 |
| 11 |
Data_Laura_3 |
| 12 |
galaxy_tools |
| 13 |
Downloads2 |
| 14 |
Downloads |
| 15 |
mom-baby_com_cn |
| 16 |
‘VirtualBox VMs2’ |
| 17 |
VirtualBox_VMs |
| 18 |
CLC_Data |
| 19 |
Work_Dir2 |
| 20 |
Work_Dir2_SGE |
| 21 |
Data_SPANDx1_Kpneumoniae_vs_Assembly1 |
| 22 |
MauveOutput |
| 23 |
Fastqs |
| 24 |
Data_Anna3_VRE_Ausbruch |
| 25 |
Work_Dir_mock_broad_mockinput |
| 26 |
Work_Dir_dM_broad_mockinput |
| 27 |
Data_Anna8_RNASeq_static_shake_deprecated |
| 28 |
PENDRIVE_cont |
| 29 |
Work_Dir_WAP_broad_mockinput |
| 30 |
Work_Dir_WAC_broad_mockinput |
| 31 |
Work_Dir_dP_broad_mockinput |
| 32 |
Data_Nicole10_16S_interlab |
| 33 |
PAPERS |
| 34 |
TB |
| 35 |
Data_Anna4_SNP |
| 36 |
Data_Carolin1_16S |
| 37 |
ChipSeq_Raw_Data3_171009_NB501882_0024_AHNGTYBGX3 |
| 38 |
m_aepfelbacher_DEL.zip |
| 39 |
Data_Anna7_RNASeq_Cytoscape |
| 40 |
Data_Nicole9_Hund_Katze_Mega |
| 41 |
Data_Anna2_CO6114 |
| 42 |
Data_Nicole3_TH17_orig |
| 43 |
Data_Nicole1_Tropheryma_whipplei |
| 44 |
results_K27 |
| 45 |
‘VirtualBox VMs’ |
| 46 |
Data_Anna6_RNASeq |
| 47 |
Data_Anna1_1585_RNAseq |
| 48 |
Data_Thaiss1_Microarray |
| 49 |
Data_Nicole7_Anelloviruses_Polyomavirus |
| 50 |
Data_Nina1_Nicole5_1-76 |
| 51 |
Data_Nina1_merged |
| 52 |
Data_Nicole8_Lamprecht |
| 53 |
Data_Anna5_SNP |
| 54 |
chipseq |
| 55 |
Downloads_DEL |
| 56 |
Data_Gagliani2_enriched_16S |
| 57 |
Data_Gagliani1_18S_16S |
| 58 |
m_aepfelbacher |
| 59 |
Data_Susanne_WGS_3amplicons |
/media/jhuang/Titisee
| # |
Name |
| 1 |
Data_Anna4_SNP |
| 2 |
Data_Anna5_SNP_rsync_error |
| 3 |
TRASH |
| 4 |
Data_Nicole6_HEV_4_SNP_calling_PE_DEL |
| 5 |
Data_Nina1_Nicole7 |
| 6 |
Data_Nicole6_HEV_4_SNP_calling_SE_DEL |
| 7 |
180119_M03701_0115_000000000-BFG46.zip |
| 8 |
Data_Nicole10_16S_interlab_PUBLISHED |
| 9 |
Anna11_assemblies |
| 10 |
Anna11_trees |
| 11 |
Data_Nicole6_HEV_new_orig_fastqs |
| 12 |
Data_Anna9_OXA-48_or_OXA-181 |
| 13 |
bengal_results_v1_2018 |
| 14 |
DO.pdf |
| 15 |
damian_DEL |
| 16 |
MAGpy_db |
| 17 |
UGENE_v1_32_data_cistrome |
| 18 |
UGENE_v1_32_data_ngs_classification |
| 19 |
Data_Nicole6_HEV_Swantje |
| 20 |
Data_Nico_Gagliani |
| 21 |
GAMOLA2_prototyp |
| 22 |
Thomas_methylation_EPIC_DO |
| 23 |
Data_Nicola_Schaltenberg |
| 24 |
Data_Nicola_Schaltenberg_PICRUSt |
| 25 |
HOME_FREIBURG |
| 26 |
Data_Francesco_16S |
| 27 |
3rd_party |
| 28 |
ConsPred_prokaryotic_genome_annotation |
| 29 |
‘System Volume Information’ |
| 30 |
damian_v201016 |
| 31 |
Data_Holger_VRE |
| 32 |
Data_Holger_Pseudomonas_aeruginosa_SNP |
| 33 |
Eigene_Ordner_HR |
| 34 |
GAMOLA2 |
| 35 |
Data_Anastasia_RNASeq |
| 36 |
Data_Amir_PUBLISHED |
| 37 |
‘$RECYCLE.BIN’ |
| 38 |
Data_Xiaobo_10x_3 |
| 39 |
Data_Tam_DNAseq_2023_Comparative_ATCC19606_AYE_ATCC17978 |
| 40 |
Data_Holger_S.epidermidis_short |
| 41 |
TEMP |
| 42 |
Data_Holger_S.epidermidis_long |
/media/jhuang/Elements(Denise_ChIPseq)
| # |
Name |
| 1 |
Data_Denise_LTtrunc_H3K27me3_2_results_DEL |
| 2 |
Data_Denise_LTtrunc_H3K4me3_2_results_DEL |
| 3 |
Data_Anna12_HAPDICS_final_not_finished_DEL |
| 4 |
m_aepfelbacher_DEL |
| 5 |
Data_Damian |
| 6 |
ST772_DEL |
| 7 |
ALL_trimmed_part_DEL |
| 8 |
Data_Denise_ChIPSeq_Protocol1 |
| 9 |
Data_Pietschmann_HCV_Amplicon |
| 10 |
Data_Nicole6_HEV_ownMethod_new |
| 11 |
HD04-1.fasta |
| 12 |
RNAHiSwitch_ |
| 13 |
RNAHiSwitch__ |
| 14 |
RNAHiSwitch___ |
| 15 |
RNAHiSwitchpaper |
| 16 |
RNAHiSwitch_milestone1_DELETED |
| 17 |
RNAHiSwitch_paper.tar.gz |
| 18 |
RNAHiSwitch_paper_DELETED |
| 19 |
RNAHiSwitch_milestone1 |
| 20 |
RNAHiSwitch_paper |
| 21 |
Ute_RNASeq_results |
| 22 |
Ute_miRNA_results_38 |
| 23 |
RNAHiSwitch |
| 24 |
Data_HepE_Freiburg_PUBLISHED |
| 25 |
Data_INTENSO_2022-06 |
| 26 |
‘$RECYCLE.BIN’ |
| 27 |
‘System Volume Information’ |
| 28 |
Data_Anna_Mixta_hanseatica_PUBLISHED |
| 29 |
coi_disclosure.docx |
| 30 |
Data_Jingang |
| 31 |
**Data_Susanne_16S_re_UNPUBLISHED *** |
| 32 |
Data_Denise_ChIPSeq_Protocol2 |
| 33 |
Data_Caroline_RNAseq_wt_timecourse |
| 34 |
Data_Caroline_RNAseq_brain_organoids |
| 35 |
Data_Amir_PUBLISHED_DEL |
| 36 |
Data_download_virus_fam |
| 37 |
Data_Gunnar_Yersiniomics_COPYFAILED_DEL |
| 38 |
Data_Paul_and_Marc_Epidome_batch3 |
| 39 |
ifconfig_hamm.txt |
| 40 |
Data_Soeren_2023_PUBLISHING |
| 41 |
Data_Birthe_Svenja_RSV_Probe3_PUBLISHING |
| 42 |
Data_Ute |
| 43 |
**Data_Susanne_16S_UNPUBLISHED *** |
/media/jhuang/Seagate Expansion Drive(HOffice)
| # |
Name |
| 1 |
SeagateExpansion.ico |
| 2 |
Autorun.inf |
| 3 |
Start_Here_Win.exe |
| 4 |
Warranty.pdf |
| 5 |
Start_Here_Mac.app |
| 6 |
Seagate |
| 7 |
HomeOffice_DIR (Data_Anna_HAPDICS_RNASeq, From_Samsung_T5) |
| 8 |
DATA_COPY_FROM_178528 (copy_and_clean.sh, logfile_jhuang.log, jhuang) |
| 9 |
‘System Volume Information’ |
| 10 |
‘$RECYCLE.BIN’ |
/media/jhuang/Elements(Anna_C.arnes)
| # |
Name |
| 1 |
Data_Swantje_HEV_using_viral-ngs |
| 2 |
VIPER_static_DEL |
| 3 |
Data_Nicole6_HEV_Swantje1_blood |
| 4 |
Data_Nicole6_HEV_benchmark |
| 5 |
Data_Denise_RNASeq_GSE79958 |
| 6 |
Data_16S_Leonie_from_Nico_Gaglianis |
| 7 |
Fastqs_19-21 |
| 8 |
‘System Volume Information’ |
| 9 |
Data_Luise_Epidome_test |
| 10 |
Data_Anna_C.acnes_PUBLISHED |
| 11 |
Data_Denise_LT_DNA_Bindung |
| 12 |
Data_Denise_LT_K331A_RNASeq |
| 13 |
Data_Luise_Epidome_batch1 |
| 14 |
Data_Luise_Pseudomonas_aeruginosa_PUBLISHED |
| 15 |
Data_Luise_Epidome_batch2 |
| 16 |
picrust2_out_2024_2 |
| 17 |
‘$RECYCLE.BIN’ |
/media/jhuang/Seagate Expansion Drive(DATA_COPY_FROM_hamburg)
| # |
Name |
| 1 |
Autorun.inf |
| 2 |
Start_Here_Win.exe |
| 3 |
Warranty.pdf |
| 4 |
Start_Here_Mac.app |
| 5 |
Seagate |
| 6 |
DATA_COPY_TRANSFER_INCOMPLETE_DEL |
| 7 |
DATA_COPY_FROM_hamburg |
/media/jhuang/Seagate Expansion Drive(Seagate_1)
| # |
Name |
| 1 |
RNA_seq_analysis_tools_2013 |
| 2 |
Data_Laura0 |
| 3 |
Data_Petra_Arck |
| 4 |
Data_Martin_mycoplasma |
| 5 |
chromhmm-enhancers |
| 6 |
ChromHMM_Dir |
| 7 |
Data_Denise_sT_H3K4me3 |
| 8 |
Data_Denise_sT_H3K27me3 |
| 9 |
Start_Here_Mac.app |
| 10 |
Seagate |
| 11 |
Data_Nicole16_parapoxvirus |
| 12 |
Project_h_rohde_Susanne_WGS_unbiased_DEL.zip |
| 13 |
Data_Denise_ChIPSeq_Protocol1 |
| 14 |
Data_ENNGS_pathogen_detection_pipeline_comparison |
| 15 |
j_huang_201904_202002 |
| 16 |
Data_Laura_ChIPseq_GSE120945 |
| 17 |
batch_200314_incomplete |
| 18 |
m_aepfelbacher.zip |
| 19 |
m_error_DEL |
| 20 |
batch_200325 |
| 21 |
batch_200319 |
| 22 |
GAMOLA2_prototyp |
| 23 |
Data_Nicola_Gagliani |
| 24 |
2017-18_raw_data |
| 25 |
Data_Arck_MeDIP |
| 26 |
trimmed |
| 27 |
Data_Nicole_16S_Christmas_2020_2 |
| 28 |
j_huang_202007_202012 |
| 29 |
Data_Nicole_16S_Christmas_2020 |
| 30 |
Downloads_2021-01-18_DEL |
| 31 |
Data_Laura_plasmid |
| 32 |
Data_Laura_16S_2_re |
| 33 |
Data_Laura_16S_2 |
| 34 |
Data_Laura_16S_2re |
| 35 |
Data_Laura_16S_merged |
| 36 |
Downloads_DEL |
| 37 |
Data_Laura_16S |
| 38 |
Data_Anna12_HAPDICS_final |
| 39 |
‘$RECYCLE.BIN’ |
| 40 |
‘System Volume Information’ |
/media/jhuang/Seagate Expansion Drive(Seagate_2)
| # |
Name |
| 1 |
Data_Nicole4_TH17 |
| 2 |
Start_Here_Win.exe |
| 3 |
Autorun.inf |
| 4 |
Warranty.pdf |
| 5 |
Start_Here_Mac.app |
| 6 |
Seagate |
| 7 |
Data_Denise_RNASeq_trimmed_DEL |
| 8 |
HD12 |
| 9 |
Qi_panGenome |
| 10 |
ALL |
| 11 |
fastq_HPI_bw_2019_08_and_2020_02 |
| 12 |
f1_R1_link.sh |
| 13 |
f1_R2_link.sh |
| 14 |
rtpd_files |
| 15 |
m_aepfelbacher.zip |
| 16 |
Data_Nicole_16S_Hamburg_Odense_Cornell_Muenster |
| 17 |
HyAsP_incomplete_genomes |
| 18 |
HyAsP_normal_sampled_input |
| 19 |
HyAsP_complete_genomes |
| 20 |
video.zip |
| 21 |
sam2bedgff.pl |
| 22 |
HD04.infection.hS_vs_HD04.nose.hS_annotated_degenes.xls |
| 23 |
ALL83 |
| 24 |
Data_Pietschmann_RSV_Probe_PUBLISHED |
| 25 |
HyAsP_normal |
| 26 |
Data_Manthey_16S |
| 27 |
rtpd_files_DEL |
| 28 |
HyAsP_bold |
| 29 |
Data_HEV |
| 30 |
Seq_VRE_hybridassembly |
| 31 |
Data_Anna12_HAPDICS_raw_data_shovill_prokka |
| 32 |
Data_Anna_HAPDICS_WGS_ALL |
| 33 |
Data_HEV_Freiburg_2020 |
| 34 |
Data_Nicole_HDV_Recombination_PUBLISHED |
| 35 |
s_hero2x |
| 36 |
201030_M03701_0207_000000000-J57B4.zip |
| 37 |
README |
| 38 |
‘README(1)’ |
| 39 |
dna2.fasta.fai |
| 40 |
91.pep |
| 41 |
91.orf |
| 42 |
91.orf.fai |
| 43 |
dgaston-dec-06-2012-121211124858-phpapp01.pdf |
| 44 |
tileshop.fcgi |
| 45 |
ppat.1009304.s016.tif |
| 46 |
sequence.txt |
| 47 |
‘sequence(1).txt’ |
| 48 |
GSE128169_series_matrix.txt.gz |
| 49 |
GSE128169_family.soft.gz |
| 50 |
Data_Anna_HAPDICS_RNASeq |
| 51 |
Data_Christopher_MeDIP_MMc_PUBLISHED |
| 52 |
Data_Gunnar_Yersiniomics_IMCOMPLETE_DEL |
| 53 |
Data_Denise_RNASeq |
| 54 |
‘System Volume Information’ |
| 55 |
‘$RECYCLE.BIN’ |
/media/jhuang/Elements(An14_RNAs)
| # |
Name |
| 1 |
Data_Anna10_RP62A |
| 2 |
Data_Nicole12_16S_Kluwe_Bunders |
| 3 |
chromhmm-enhancers |
| 4 |
Data_Denise_sT_Methylation |
| 5 |
Data_Denise_LTtrunc_Methylation |
| 6 |
Data_16S_arckNov |
| 7 |
Data_Tabea_RNASeq |
| 8 |
nr_gz_README |
| 9 |
j_huang_raw_fq |
| 10 |
‘System Volume Information’ |
| 11 |
‘$RECYCLE.BIN’ |
| 12 |
host_refs |
| 13 |
Vraw |
| 14 |
**Data_Susanne_Amplicon_RdRp_orf1_2 *** |
| 15 |
tmp |
| 16 |
Data_RNA188_Paul_Becher |
| 17 |
Data_ChIPSeq_Laura |
| 18 |
Data_16S_arckNov_review_PUBLISHED |
| 19 |
Data_16S_arckNov_re |
| 20 |
Fastqs |
| 21 |
Data_Tabea_RNASeq_submission |
| 22 |
Data_Anna_Cutibacterium_acnes_DEL |
| 23 |
Data_Silvia_RNASeq_SUBMISSION |
| 24 |
Data_Hannes_ChIPSeq |
| 25 |
Data_Anna14_RNASeq_to_be_DEL |
| 26 |
Data_Pietschmann_RSV_Probe2_PUBLISHED |
| 27 |
Data_Holger_Klebsiella_pneumoniae_SNP_PUBLISHING |
| 28 |
Data_Anna14_RNASeq_plus_public |
/media/jhuang/Elements(Indra_HAPDICS)
| # |
Name |
| 1 |
Data_Anna11_Sepdermidis_DEL |
| 2 |
HD15_without_10 |
| 3 |
HD31 |
| 4 |
HD33 |
| 5 |
HD39 |
| 6 |
HD43 |
| 7 |
HD46 |
| 8 |
HD15_with_10 |
| 9 |
HD26 |
| 10 |
HD59 |
| 11 |
HD25 |
| 12 |
HD21 |
| 13 |
HD17 |
| 14 |
HD04 |
| 15 |
Data_Anna11_Pair1-6_P6 |
| 16 |
Data_Anna12_HAPDICS_HyAsP |
| 17 |
HAPDICS_hyasp_plasmids |
| 18 |
Data_Anna_HAPDICS_review |
| 19 |
data_overview.txt |
| 20 |
align_assem_res_DEL |
| 21 |
‘System Volume Information’ |
| 22 |
EXCHANGE_DEL |
| 23 |
Data_Indra_H3K4me3_public |
| 24 |
Data_Gunnar_MS |
| 25 |
‘$RECYCLE.BIN’ |
| 26 |
UKE_DELLWorkstation_C_Users_indbe_Desktop |
| 27 |
Linux_DELLWorkstation_C_Users_indbe_VirtualBoxVMs |
| 28 |
Data_Anna_HAPDICS_RNASeq_rawdata |
| 29 |
Data_Indra_H3K27ac_public |
| 30 |
Data_Holger_Klebsiella_pneumoniae_SNP_PUBLISHING |
| 31 |
DATA_INDRA_RNASEQ |
| 32 |
DATA_INDRA_CHIPSEQ |
/media/jhuang/Elements(jhuang_*)
| # |
Name |
| 1 |
‘Install Western Digital Software for Windows.exe’ |
| 2 |
‘Install Western Digital Software for Mac.dmg’ |
| 3 |
‘System Volume Information’ |
| 4 |
‘$RECYCLE.BIN’ |
| 5 |
20250203_FS10003086_95_BTR67811-0621 |
/media/jhuang/Smarty
| # |
Name |
| 1 |
lost+found |
| 2 |
Blast_db |
| 3 |
temporary_files_DEL |
| 4 |
ALIGN_ASSEM |
| 5 |
Data_Paul_Staphylococcus_epidermidis |
| 6 |
Data_16S_Degenhardt_Marius_DEL |
| 7 |
Data_Gunnar_Yersiniomics_DEL |
| 8 |
Data_Manja_RNAseq_Organoids_Virus |
| 9 |
Data_Emilia_MeDIP |
| 10 |
DjangoApp_Backup_2023-10-30 |
| 11 |
ref |
| 12 |
Data_Michelle_RNAseq_2025_raw_data_DEL_AFTER_UPLOAD_GEO |
Original input (as one point)
/media/jhuang/INTENSO is empty --> Now the data are on ~/DATA_Intenso
/dev/sdg1 3,7T 512K 3,7T 1% /media/jhuang/INTENSO
jhuang@WS-2290C:~/DATA$ ls -tlrh
total 1,6M
drwxrwxrwx 6 jhuang jhuang 4,0K Okt 26 2022 Data_Ute_MKL1
drwxrwxrwx 8 jhuang jhuang 4,0K Jan 13 2023 Data_Ute_RNA_4_2022-11_test
drwxrwxr-x 7 jhuang jhuang 4,0K Mär 8 2023 Data_Ute_RNA_3
drwxr-xr-x 11 jhuang jhuang 4,0K Dez 19 2023 Data_Susanne_Carotis_RNASeq_PUBLISHING
drwxr-xr-x 21 jhuang jhuang 4,0K Jun 18 2024 Data_Jiline_Yersinia_SNP
drwxrwxr-x 5 jhuang jhuang 4,0K Jul 22 2024 Data_Tam_ABAYE_RS05070_on_A_calcoaceticus_baumannii_complex_DUPLICATED_DEL
drwxr-xr-x 2 jhuang jhuang 4,0K Jul 23 2024 Data_Nicole_CRC1648
drwxr-xr-x 4 jhuang jhuang 4,0K Sep 6 2024 Mouse_HS3ST1_12373_out
drwxr-xr-x 4 jhuang jhuang 4,0K Sep 6 2024 Mouse_HS3ST1_12175_out
drwxrwxr-x 10 jhuang jhuang 4,0K Sep 12 2024 Data_Biobakery
drwxrwxr-x 6 jhuang jhuang 4,0K Sep 23 2024 Data_Xiaobo_10x_2
drwxr-xr-x 4 jhuang jhuang 4,0K Sep 23 2024 Data_Xiaobo_10x_3
drwxr-xr-x 3 jhuang jhuang 4,0K Sep 26 2024 Talk_Nicole_CRC1648
drwxr-xr-x 2 jhuang jhuang 4,0K Sep 26 2024 Talks_Bioinformatics_Meeting
drwxr-xr-x 2 jhuang jhuang 12K Sep 26 2024 Talks_resources
drwxrwxr-x 6 jhuang jhuang 12K Okt 10 2024 Data_Susanne_MPox_DAMIAN
drwxrwxr-x 3 jhuang jhuang 4,0K Okt 14 2024 Data_host_transcriptional_response
drwxr-xr-x 13 jhuang jhuang 4,0K Okt 23 2024 Talks_including_DEEP-DV
drwxrwxr-x 2 jhuang jhuang 4,0K Okt 24 2024 DOKTORARBEIT
drwxrwxr-x 18 jhuang jhuang 4,0K Nov 11 2024 Data_Susanne_MPox
drwxrwxr-x 25 jhuang jhuang 12K Nov 11 2024 Data_Jiline_Transposon
drwxrwxr-x 16 jhuang jhuang 20K Nov 25 2024 Data_Jiline_Transposon2
drwxrwxr-x 3 jhuang jhuang 4,0K Dez 13 2024 Data_Matlab
drwxrwxr-x 5 jhuang jhuang 4,0K Jan 28 2025 deepseek-ai
drwx------ 4 jhuang jhuang 4,0K Feb 5 2025 Stick_Mi_DEL
-rw-rw-r-- 1 jhuang jhuang 1,1K Feb 18 2025 TODO_shares
drwxrwxrwx 13 jhuang jhuang 4,0K Mär 3 2025 Data_Ute_RNA_4
drwxrwxr-x 2 jhuang jhuang 4,0K Mär 31 2025 Data_Liu_PCA_plot
-rw-rw-r-- 1 jhuang jhuang 43K Apr 3 2025 README_run_viral-ngs_inside_Docker
-rw-rw-r-- 1 jhuang jhuang 8,7K Apr 9 2025 README_compare_genomes
-rw-rw-r-- 1 jhuang jhuang 0 Apr 11 2025 mapped.bam
drwxrwxr-x 3 jhuang jhuang 4,0K Apr 24 2025 Data_Serpapi
drwxrwxrwx 22 jhuang jhuang 4,0K Apr 30 2025 Data_Ute_RNA_1_2
drwxrwxr-x 15 jhuang jhuang 4,0K Apr 30 2025 Data_Marc_RNAseq_2024
drwxrwxr-x 45 jhuang jhuang 12K Mai 15 2025 Data_Nicole_CaptureProbeSequencing
-rw-rw-r-- 1 jhuang jhuang 657 Mai 23 2025 LOG_mapping
drwxrwxr-x 46 jhuang jhuang 4,0K Mai 26 2025 Data_Huang_Human_herpesvirus_3
drwxrwxr-x 8 jhuang jhuang 4,0K Jun 13 2025 Data_Nicole_DAMIAN_Post-processing_Pathoprobe_FluB_Links
lrwxrwxrwx 1 jhuang jhuang 37 Jun 16 2025 Access_to_Win7 -> ./Data_Marius_16S/picrust2_out_2024_2
drwxrwxr-x 17 jhuang jhuang 4,0K Jun 18 2025 Data_DAMIAN_Post-processing_Flavivirus_and_FSME_and_Haemophilus
drwxr-xr-x 42 jhuang jhuang 36K Jun 23 2025 Data_Luise_Sepi_STKN
drwxrwxr-x 29 jhuang jhuang 20K Jul 22 2025 Data_Patricia_Sepi_7samples
drwxr-xr-x 9 jhuang jhuang 4,0K Aug 8 2025 Data_Soeren_2025_PUBLISHING
drwxrwxr-x 9 jhuang jhuang 4,0K Aug 13 2025 Data_Ben_RNAseq_2025
drwxrwxr-x 34 jhuang jhuang 12K Sep 3 12:18 Data_Tam_DNAseq_2025_AYE-WT_Q_S_craA-Tig4_craA-1-Cm200_craA-2-Cm200
drwxrwxr-x 50 jhuang jhuang 16K Okt 6 17:59 Data_Patricia_Transposon
drwxrwxr-x 23 jhuang jhuang 12K Okt 20 13:27 Data_Patricia_Transposon_2025
drwxrwxr-x 2 jhuang jhuang 4,0K Okt 23 12:21 Colocation_Space
drwxrwxr-x 2 jhuang jhuang 4,0K Okt 27 12:56 Data_Tam_Methylation_2025_empty
-rw-rw-r-- 1 jhuang jhuang 151K Nov 3 13:01 2025-11-03_eVB-Schreiben_12-57.pdf
-rw-rw-r-- 1 jhuang jhuang 67K Nov 5 16:59 DEGs_Group1_A1-A3+A8-A10_vs_Group2_B10-B16.png
-rw-rw-r-- 1 jhuang jhuang 687K Nov 14 09:55 README.pdf
drwxrwxr-x 2 jhuang jhuang 4,0K Nov 24 15:43 Data_Hannes_JCM00612
drwxrwxr-x 3 jhuang jhuang 4,0K Dez 4 17:03 167_redundant_DEL
drwxrwxr-x 2 jhuang jhuang 4,0K Dez 8 10:33 Lehre_Bioinformatik
drwxrwxr-x 27 jhuang jhuang 12K Dez 8 11:29 Data_Ben_Boruta_Analysis
drwxrwxr-x 18 jhuang jhuang 4,0K Dez 8 17:39 Data_Childrensclinic_16S_2025_DEL
drwxrwxr-x 2 jhuang jhuang 4,0K Dez 10 10:05 Data_Ben_Mycobacterium_pseudoscrofulaceum
-rw-rw-r-- 1 jhuang jhuang 8,9K Dez 15 12:42 Foong_RNA_mSystems_Huang_Changed.txt
drwxrwxr-x 22 jhuang jhuang 4,0K Dez 17 13:07 Data_Pietro_Scatturo_and_Charlotte_Uetrecht_16S_2025
drwxrwxr-x 8 jhuang jhuang 4,0K Dez 18 10:45 Data_JuliaBerger_RNASeq_SARS-CoV-2
drwxrwxr-x 19 jhuang jhuang 4,0K Jan 3 17:42 Data_PaulBongarts_S.epidermidis_HDRNA
lrwxrwxrwx 1 jhuang jhuang 31 Jan 12 14:30 Data_Ute -> /media/jhuang/Elements/Data_Ute
drwxrwxr-x 12 jhuang jhuang 4,0K Jan 16 12:44 Data_Foong_DNAseq_2025_AYE_Dark_vs_Light_TODO
drwxrwxrwx 22 jhuang jhuang 4,0K Jan 16 12:48 Data_Foong_RNAseq_2021_ATCC19606_Cm
drwxrwxr-x 2 jhuang jhuang 4,0K Jan 16 13:02 Data_Tam_Funding
drwxrwxr-x 9 jhuang jhuang 4,0K Jan 16 13:32 Data_Tam_RNAseq_2025_LB-AB_IJ_W1_Y1_WT_vs_Mac-AB_IJ_W1_Y1_WT_on_ATCC19606
drwxrwxr-x 12 jhuang jhuang 4,0K Jan 16 13:32 Data_Tam_RNAseq_2025_subMIC_exposure_on_ATCC19606
-rw-rw-r-- 1 jhuang jhuang 1,2K Jan 16 13:34 Data_Tam.txt
drwxrwxr-x 16 jhuang jhuang 4,0K Jan 16 13:37 Data_Tam_RNAseq_2024_AUM_MHB_Urine_on_ATCC19606
drwxrwxr-x 10 jhuang jhuang 4,0K Jan 16 18:22 Data_Tam_Metagenomics_2026
drwxrwxr-x 6 jhuang jhuang 16K Jan 23 16:35 Data_Michelle
drwxrwxr-x 38 jhuang jhuang 12K Jan 28 15:20 Data_Nicole_16S_2025_Childrensclinic
drwxr-xr-x 145 jhuang jhuang 36K Jan 29 10:49 Data_Sophie_HDV_Sequences
drwxrwxr-x 4 jhuang jhuang 4,0K Jan 30 11:44 Data_Tam_DNAseq_2026_19606deltaIJfluE
-rw-rw-r-- 1 jhuang jhuang 63K Jan 30 17:53 README_nf-core
drwxrwxr-x 22 jhuang jhuang 4,0K Feb 4 10:43 Data_Vero_Kymographs
drwxrwxr-x 13 jhuang jhuang 4,0K Feb 4 14:06 Access_to_Win10
drwxrwxr-x 7 jhuang jhuang 4,0K Feb 5 11:59 Data_Patricia_AMRFinderPlus_2025
drwxrwxr-x 45 jhuang jhuang 4,0K Feb 6 11:54 Data_Tam_DNAseq_2025_Unknown-adeABadeIJ_adeIJK_CM1_CM2
drwxrwxr-x 41 jhuang jhuang 12K Feb 9 15:11 Data_Damian
drwxrwxr-x 6 jhuang jhuang 4,0K Feb 13 12:48 Data_Karoline_16S
drwxrwxr-x 13 jhuang jhuang 12K Feb 13 18:09 Data_JuliaFuchs_RNAseq_2025
drwxrwxr-x 18 jhuang jhuang 4,0K Feb 16 11:19 Data_Tam_DNAseq_2025_ATCC19606-Y1Y2Y3Y4W1W2W3W4_TODO
drwxrwxr-x 34 jhuang jhuang 4,0K Feb 16 15:54 Data_Tam_DNAseq_2026_Acinetobacter_harbinensis
drwxrwxr-x 19 jhuang jhuang 4,0K Feb 16 17:13 Data_Benjamin_DNAseq_2026_GE11174
drwxrwxrwx 36 jhuang jhuang 12K Feb 17 15:02 Data_Susanne_spatialRNA_2022.9.1_backup
drwxrwxr-x 39 jhuang jhuang 12K Feb 17 15:12 Data_Susanne_spatialRNA
jhuang@WS-2290C:~/DATA_A$ ls -ltrh
total 24K
drwxr-xr-x 7 jhuang jhuang 4,0K Jun 18 2024 Data_Damian_NEW_CREATED
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_R_bubbleplots
drwxr-xr-x 16 jhuang jhuang 4,0K Jun 18 2024 Data_Ute_TRANSFERED_DEL
drwxr-xr-x 2 jhuang jhuang 4,0K Okt 7 2024 Paper_Target_capture_sequencing_MHH_PUBLISHED
drwxr-xr-x 20 jhuang jhuang 4,0K Okt 8 2024 Data_Nicole8_Lamprecht_new_PUBLISHED
drwxrwxrwx 8 jhuang jhuang 4,0K Mai 21 2025 Data_Samira_RNAseq
jhuang@WS-2290C:~/DATA_B$ ls -tlrh
total 136K
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_DAMIAN_endocarditis_encephalitis
drwxr-xr-x 8 jhuang jhuang 4,0K Jun 18 2024 Data_Denise_sT_PUBLISHING
drwxr-xr-x 12 jhuang jhuang 4,0K Jun 18 2024 Data_Fran2_16S_func
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Holger_5179-R1_vs_5179
drwxr-xr-x 16 jhuang jhuang 4,0K Jun 18 2024 Antraege_
drwxr-xr-x 18 jhuang jhuang 4,0K Jun 18 2024 Data_16S_Nicole_210222
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 Data_Adam_Influenza_A_virus
drwxr-xr-x 14 jhuang jhuang 12K Jun 18 2024 Data_Anna_Efaecium_assembly
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Bactopia
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Data_Ben_RNAseq
drwxr-xr-x 7 jhuang jhuang 4,0K Jun 18 2024 Data_Johannes_PIV3
drwxr-xr-x 19 jhuang jhuang 4,0K Jun 18 2024 Data_Luise_Epidome_longitudinal_nose
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 Data_Manja_Hannes_Probedesign
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Marc_AD_PUBLISHING
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Marc_RNA-seq_Saureus_Review
drwxr-xr-x 17 jhuang jhuang 4,0K Jun 18 2024 Data_Nicole_16S
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_Nicole_cfDNA_pathogens
drwxr-xr-x 16 jhuang jhuang 4,0K Jun 18 2024 Data_Ring_and_CSF_PegivirusC_DAMIAN
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_Song_Microarray
drwxr-xr-x 11 jhuang jhuang 4,0K Jun 18 2024 Data_Susanne_Omnikron
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_Viro
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Doktorarbeit
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Poster_Rohde_20230724
drwxr-xr-x 6 jhuang jhuang 4,0K Jul 12 2024 Data_Django
drwxr-xr-x 35 jhuang jhuang 4,0K Okt 21 2024 Data_Holger_S.epidermidis_1585_5179_HD05
drwxr-xr-x 9 jhuang jhuang 4,0K Nov 18 2024 Data_Manja_RNAseq_Organoids_Virus
drwxr-xr-x 2 jhuang jhuang 4,0K Feb 21 2025 Data_Holger_MT880870_MT880872_Annotation
drwxr-xr-x 12 jhuang jhuang 4,0K Apr 8 2025 Data_Soeren_RNA-seq_2022
drwxr-xr-x 5 jhuang jhuang 4,0K Apr 11 2025 Data_Manja_RNAseq_Organoids_Merged
drwxr-xr-x 24 jhuang jhuang 4,0K Apr 25 2025 Data_Gunnar_Yersiniomics
drwxr-xr-x 10 jhuang jhuang 4,0K Jan 16 17:14 Data_Manja_RNAseq_Organoids
drwxr-xr-x 3 jhuang jhuang 4,0K Feb 17 12:11 Data_Susanne_Carotis_MS
jhuang@WS-2290C:~/DATA_C$ ls -tlrh
total 13G
-rwxr-xr-x 1 jhuang jhuang 1,7M Jun 18 2024 2022-10-27_IRI_manuscript_v03_JH.docx
-rwxr-xr-x 1 jhuang jhuang 7,1K Jun 18 2024 16304905.fasta
-rwxr-xr-x 1 jhuang jhuang 55K Jun 18 2024 '16S data manuscript_NF.docx'
-rwxr-xr-x 1 jhuang jhuang 792K Jun 18 2024 180820_2_supp_4265595_sw6zjk.docx
-rwxr-xr-x 1 jhuang jhuang 17K Jun 18 2024 180820_2_supp_4265596_sw6zjk.docx
-rwxr-xr-x 1 jhuang jhuang 12K Jun 18 2024 1a_vs_3.csv
-rwxr-xr-x 1 jhuang jhuang 90K Jun 18 2024 '2.05.01.05-A01 Urlaubsantrag-Shuting-beantragt.pdf'
-rwxr-xr-x 1 jhuang jhuang 708K Jun 18 2024 2014SawickaBBA.pdf
-rwxr-xr-x 1 jhuang jhuang 61K Jun 18 2024 20160509Manuscript_NDM_OXA_mitKomm.doc
-rwxr-xr-x 1 jhuang jhuang 289K Jun 18 2024 220607_Agenda_monthly_meeting.pdf
-rwxr-xr-x 1 jhuang jhuang 14K Jun 18 2024 '20221129 Table mutations.docx'
-rwxr-xr-x 1 jhuang jhuang 12G Jun 18 2024 230602_NB501882_0428_AHKG53BGXT.zip
-rwxr-xr-x 1 jhuang jhuang 107K Jun 18 2024 362383173.rar
-rwxr-xr-x 1 jhuang jhuang 128K Jun 18 2024 562.9459.1.fa
-rwxr-xr-x 1 jhuang jhuang 126K Jun 18 2024 562.9459.1_rc.fa
-rwxr-xr-x 1 jhuang jhuang 1,6M Jun 18 2024 ASA3P.pdf
-rwxr-xr-x 1 jhuang jhuang 21K Jun 18 2024 All_indels_annotated_vHR.xlsx
-rwxr-xr-x 1 jhuang jhuang 11K Jun 18 2024 'Amplikon_indeces_Susanne +groups.xlsx'
-rwxr-xr-x 1 jhuang jhuang 9,6K Jun 18 2024 Amplikon_indeces_Susanne.xlsx
-rwxr-xr-x 1 jhuang jhuang 68 Jun 18 2024 GAMOLA2
-rwxr-xr-x 1 jhuang jhuang 88 Jun 18 2024 Data_Susanne_Carotis_spatialRNA_PUBLISHING
-rwxr-xr-x 1 jhuang jhuang 112 Jun 18 2024 Data_Paul_Staphylococcus_epidermidis
-rwxr-xr-x 1 jhuang jhuang 118 Jun 18 2024 Data_Nicola_Schaltenberg_PICRUSt
-rwxr-xr-x 1 jhuang jhuang 100 Jun 18 2024 Data_Nicola_Schaltenberg
-rwxr-xr-x 1 jhuang jhuang 94 Jun 18 2024 Data_Nicola_Gagliani
-rwxr-xr-x 1 jhuang jhuang 96 Jun 18 2024 Data_methylome_MMc
-rwxr-xr-x 1 jhuang jhuang 78 Jun 18 2024 Data_Jingang
-rwxr-xr-x 1 jhuang jhuang 112 Jun 18 2024 Data_Indra_RNASeq_GSM2262901
-rwxr-xr-x 1 jhuang jhuang 84 Jun 18 2024 Data_Holger_VRE
-rwxr-xr-x 1 jhuang jhuang 128 Jun 18 2024 Data_Holger_Pseudomonas_aeruginosa_SNP
-rwxr-xr-x 1 jhuang jhuang 92 Jun 18 2024 Data_Hannes_ChIPSeq
-rwxr-xr-x 1 jhuang jhuang 76 Jun 18 2024 Data_Emilia_MeDIP
-rwxr-xr-x 1 jhuang jhuang 88 Jun 18 2024 Data_ChristophFR_HepE_published
-rwxr-xr-x 1 jhuang jhuang 158 Jun 18 2024 Data_Christopher_MeDIP_MMc_published
-rwxr-xr-x 1 jhuang jhuang 104 Jun 18 2024 Data_Anna_Kieler_Sepi_Staemme
-rwxr-xr-x 1 jhuang jhuang 136 Jun 18 2024 Data_Anna12_HAPDICS_final
-rwxr-xr-x 1 jhuang jhuang 96 Jun 18 2024 Data_Anastasia_RNASeq_PUBLISHING
-rwxr-xr-x 1 jhuang jhuang 169K Jun 18 2024 Aufnahmeantrag_komplett_10_2022.pdf
-rwxr-xr-x 1 jhuang jhuang 1,2M Jun 18 2024 Astrovirus.pdf
-rwxr-xr-x 1 jhuang jhuang 732 Jun 18 2024 COMMANDS
-rwxr-xr-x 1 jhuang jhuang 690 Jun 18 2024 Bacterial_pipelines.txt
-rwxr-xr-x 1 jhuang jhuang 16M Jun 18 2024 COMPSRA_uke_DEL.jar
-rwxr-xr-x 1 jhuang jhuang 239K Jun 18 2024 ChIPSeq_pipeline_desc.docx
-rwxr-xr-x 1 jhuang jhuang 385K Jun 18 2024 ChIPSeq_pipeline_desc.pdf
-rwxr-xr-x 1 jhuang jhuang 2,1M Jun 18 2024 Comparative_genomic_analysis_of_eight_novel_haloal.pdf
-rwxr-xr-x 1 jhuang jhuang 64K Jun 18 2024 CvO_Klassenliste_7_3.pdf
-rwxr-xr-x 1 jhuang jhuang 649K Jun 18 2024 'Copy of pool_b1_CGATGT_300.xlsx'
-rwxr-xr-x 1 jhuang jhuang 3,9K Jun 18 2024 Fran_16S_Exp8-17-21-27.txt
-rwxr-xr-x 1 jhuang jhuang 463 Jun 18 2024 HPI_DRIVE
-rwxr-xr-x 1 jhuang jhuang 179K Jun 18 2024 HEV_aligned.fasta
-rwxr-xr-x 1 jhuang jhuang 4,1K Jun 18 2024 INTENSO_DIR
-rwxr-xr-x 1 jhuang jhuang 14K Jun 18 2024 HPI_samples_for_NGS_29.09.22.xlsx
-rwxr-xr-x 1 jhuang jhuang 4,3K Jun 18 2024 Hotmail_to_Gmail
-rwxr-xr-x 1 jhuang jhuang 13M Jun 18 2024 Indra_Thesis_161020.pdf
-rwxr-xr-x 1 jhuang jhuang 5,2K Jun 18 2024 'LT K331A.gbk'
-rwxr-xr-x 1 jhuang jhuang 0 Jun 18 2024 LOG_p954_stat
-rwxr-xr-x 1 jhuang jhuang 684K Jun 18 2024 LOG
-rwxr-xr-x 1 jhuang jhuang 197K Jun 18 2024 Manuscript_10_02_2021.docx
-rwxr-xr-x 1 jhuang jhuang 595K Jun 18 2024 Metagenomics_Tools_and_Insights.pdf
-rwxr-xr-x 1 jhuang jhuang 14K Jun 18 2024 'Miseq Amplikon LAuf April.xlsx'
-rwxr-xr-x 1 jhuang jhuang 2,2M Jun 18 2024 NGS.tar.gz
-rwxr-xr-x 1 jhuang jhuang 586K Jun 18 2024 Nachweis_Bakterien_Viren_im_Hochdurchsatz.pdf
-rwxr-xr-x 1 jhuang jhuang 1,2K Jun 18 2024 Nicole8_Lamprecht_logs
-rwxr-xr-x 1 jhuang jhuang 24M Jun 18 2024 Nanopore.handouts.pdf
-rwxr-xr-x 1 jhuang jhuang 113K Jun 18 2024 'Norovirus paper Susanne 191105.docx'
-rwxr-xr-x 1 jhuang jhuang 503K Jun 18 2024 PhyloRNAalifold.pdf
-rwxr-xr-x 1 jhuang jhuang 19K Jun 18 2024 README_R
-rwxr-xr-x 1 jhuang jhuang 137K Jun 18 2024 README_RNAHiSwitch_DEL
-rwxr-xr-x 1 jhuang jhuang 8,3M Jun 18 2024 RNA-NGS_Analysis_modul3_NanoStringNorm.zip
-rwxr-xr-x 1 jhuang jhuang 57K Jun 18 2024 RNAConSLOptV1.2.tar.gz
-rwxr-xr-x 1 jhuang jhuang 17K Jun 18 2024 'RSV GFP5 including 3`UTR.docx'
-rwxr-xr-x 1 jhuang jhuang 238 Jun 18 2024 SNPs_on_pangenome.txt
-rwxr-xr-x 1 jhuang jhuang 55 Jun 18 2024 SERVER
-rwxr-xr-x 1 jhuang jhuang 26M Jun 18 2024 R_tutorials-master.zip
-rwxr-xr-x 1 jhuang jhuang 182K Jun 18 2024 Rawdata_Readme.pdf
-rwxr-xr-x 1 jhuang jhuang 40K Jun 18 2024 SUB10826945_record_preview.txt
-rwxr-xr-x 1 jhuang jhuang 283K Jun 18 2024 S_staphylococcus_annotated_diff_expr.xls
-rwxr-xr-x 1 jhuang jhuang 2,0K Jun 18 2024 Snakefile_list
-rwxr-xr-x 1 jhuang jhuang 160K Jun 18 2024 Source_Classification_Code.rds
-rwxr-xr-x 1 jhuang jhuang 61K Jun 18 2024 Supplementary_Table_S3.xlsx
-rwxr-xr-x 1 jhuang jhuang 617 Jun 18 2024 Untitled.ipynb
-rwxr-xr-x 1 jhuang jhuang 127M Jun 18 2024 UniproUGENE_UserManual.pdf
-rwxr-xr-x 1 jhuang jhuang 14M Jun 18 2024 Untitled1.ipynb
-rwxr-xr-x 1 jhuang jhuang 110K Jun 18 2024 Untitled2.ipynb
-rwxr-xr-x 1 jhuang jhuang 2,9K Jun 18 2024 Untitled3.ipynb
-rwxr-xr-x 1 jhuang jhuang 18K Jun 18 2024 WAC6h_vs_WAP6h_down.txt
-rwxr-xr-x 1 jhuang jhuang 100 Jun 18 2024 damian_nodbs
-rwxr-xr-x 1 jhuang jhuang 45K Jun 18 2024 WAC6h_vs_WAP6h_up.txt
-rwxr-xr-x 1 jhuang jhuang 635K Jun 18 2024 'add. Figures Hamburg_UKE.pptx'
-rwxr-xr-x 1 jhuang jhuang 3,7M Jun 18 2024 all_gene_counts_with_annotation.xlsx
-rwxr-xr-x 1 jhuang jhuang 22K Jun 18 2024 app_flask.py
-rwxr-xr-x 1 jhuang jhuang 1,8K Jun 18 2024 bengal-bay-0.1.json
-rwxr-xr-x 1 jhuang jhuang 16K Jun 18 2024 bengal3_ac3.yml
-rwxr-xr-x 1 jhuang jhuang 246K Jun 18 2024 call_shell_from_Ruby.png
-rwxr-xr-x 1 jhuang jhuang 8,1K Jun 18 2024 bengal3_ac3_.yml
-rwxr-xr-x 1 jhuang jhuang 12 Jun 18 2024 empty.fasta
-rwxr-xr-x 1 jhuang jhuang 32K Jun 18 2024 coefficients_csaw_vs_diffreps.xlsx
-rwxr-xr-x 1 jhuang jhuang 4,3K Jun 18 2024 exchange.txt
-rwxr-xr-x 1 jhuang jhuang 30M Jun 18 2024 exdata-data-NEI_data.zip
-rwxr-xr-x 1 jhuang jhuang 6,6K Jun 18 2024 genes_wac6_wap6.xls
-rwxr-xr-x 1 jhuang jhuang 115M Jun 18 2024 go1.13.linux-amd64.tar.gz.1
-rwxr-xr-x 1 jhuang jhuang 29K Jun 18 2024 hev_p2-p5.fa
-rwxr-xr-x 1 jhuang jhuang 3,8K Jun 18 2024 map_corrected_backup.txt
-rwxr-xr-x 1 jhuang jhuang 325 Jun 18 2024 install_nginx_on_hamm
-rwxr-xr-x 1 jhuang jhuang 20M Jun 18 2024 hg19.rmsk.bed
-rwxr-xr-x 1 jhuang jhuang 107K Jun 18 2024 metadata-9563675-processed-ok.tsv
-rwxr-xr-x 1 jhuang jhuang 288K Jun 18 2024 mkg_sprechstundenflyer_ver1b_dezember_2019.pdf
-rwxr-xr-x 1 jhuang jhuang 588 Jun 18 2024 multiqc_config.yaml
-rwxr-xr-x 1 jhuang jhuang 38K Jun 18 2024 p11326_OMIKRON3398_corsurv.gb
-rwxr-xr-x 1 jhuang jhuang 30K Jun 18 2024 p11326_OMIKRON3398_corsurv.gb_converted.fna
-rwxr-xr-x 1 jhuang jhuang 3,9K Jun 18 2024 parseGenbank_reformat.py
-rwxr-xr-x 1 jhuang jhuang 222K Jun 18 2024 pangenome-snakemake-master.zip
-rwxr-xr-x 1 jhuang jhuang 283K Jun 18 2024 'phylo tree draft.pdf'
-rwxr-xr-x 1 jhuang jhuang 125 Jun 18 2024 qiime_params.txt
-rwxr-xr-x 1 jhuang jhuang 2,3M Jun 18 2024 pool_b1_CGATGT_300.zip
-rwxr-xr-x 1 jhuang jhuang 5,5K Jun 18 2024 qiime_params_backup.txt
-rwxr-xr-x 1 jhuang jhuang 4,5K Jun 18 2024 qiime_params_s16_s18.txt
-rwxr-xr-x 1 jhuang jhuang 68 Jun 18 2024 snakePipes
-rwxr-xr-x 1 jhuang jhuang 25K Jun 18 2024 results_description.html
-rwxr-xr-x 1 jhuang jhuang 139M Jun 18 2024 rnaalihishapes.tar.gz
-rwxr-xr-x 1 jhuang jhuang 3,4M Jun 18 2024 rnaseq_length_bias.pdf
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 3932-Leber
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 BioPython
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Biopython
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 DEEP-DV
drwxr-xr-x 13 jhuang jhuang 4,0K Jun 18 2024 DOKTORARBEIT
drwxr-xr-x 17 jhuang jhuang 4,0K Jun 18 2024 Data_16S_Arck_vaginal_stool
drwxr-xr-x 22 jhuang jhuang 4,0K Jun 18 2024 Data_16S_BS052
drwxr-xr-x 13 jhuang jhuang 4,0K Jun 18 2024 Data_16S_Birgit
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_16S_Christner
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 Data_16S_Leonie
drwxr-xr-x 11 jhuang jhuang 4,0K Jun 18 2024 Data_16S_PatientA-G_CSF
drwxr-xr-x 14 jhuang jhuang 4,0K Jun 18 2024 Data_16S_Schaltenberg
drwxr-xr-x 7 jhuang jhuang 4,0K Jun 18 2024 Data_16S_benchmark
drwxr-xr-x 7 jhuang jhuang 4,0K Jun 18 2024 Data_16S_benchmark2
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_16S_gcdh_BKV
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Alex1_Amplicon
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Alex1_SNP
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Data_Analysis_for_Life_Science
drwxr-xr-x 19 jhuang jhuang 4,0K Jun 18 2024 Data_Anna13_vanA-Element
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Anna14_PACBIO_methylation
drwxr-xr-x 8 jhuang jhuang 4,0K Jun 18 2024 Data_Anna_C.acnes2_old_DEL
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Anna_MT880872_update
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Anna_gap_filling_agrC
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_Baechlein_Hepacivirus_2018
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Bornavirus
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_CSF
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 Data_Christine_cz19-178-rothirsch-bovines-hepacivirus
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_Daniela_adenovirus_WGS
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_Emilia_MeDIP_DEL
drwxr-xr-x 14 jhuang jhuang 4,0K Jun 18 2024 Data_Francesco2021_16S
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 Data_Francesco2021_16S_re
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_Gunnar_MS
drwxr-xr-x 10 jhuang jhuang 4,0K Jun 18 2024 Data_Hannes_RNASeq
drwxr-xr-x 29 jhuang jhuang 4,0K Jun 18 2024 Data_Holger_Efaecium_variants_PUBLISHED
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Data_Holger_VRE_DEL
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_Icebear_Damian
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Indra3_H3K4_p2_DEL
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_Indra6_RNASeq_ChipSeq_Integration_DEL
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Indra_Figures
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_KatjaGiersch_new_HDV
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_MHH_Encephalitits_DAMIAN
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 Data_Manja_RPAChIPSeq_public
drwxr-xr-x 72 jhuang jhuang 12K Jun 18 2024 Data_Manuel_WGS_Yersinia
drwxr-xr-x 32 jhuang jhuang 4,0K Jun 18 2024 Data_Manuel_WGS_Yersinia2_DEL
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_Manuel_WGS_Yersinia_DEL
drwxr-xr-x 13 jhuang jhuang 4,0K Jun 18 2024 Data_Marcus_tracrRNA_structures
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Data_Mausmaki_Damian
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_Nicole1_Tropheryma_whipplei
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Nicole5
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 Data_Nicole5_77-92
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_PaulBecher_Rotavirus
drwxr-xr-x 21 jhuang jhuang 4,0K Jun 18 2024 Data_Pietschmann_HCV_Amplicon_bigFile
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Piscine_Orthoreovirus_3_in_Brown_Trout
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Proteomics
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_RNABioinformatics
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_RNAKinetics
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_R_courses
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Data_SARS-CoV-2
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 Data_SARS-CoV-2_Genome_Announcement_PUBLISHED
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Seite
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Song_aggregate_sum
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_Susanne_Amplicon_RdRp_orf1_2_re
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Tabea_RNASeq
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Data_Thaiss1_Microarray_new
drwxr-xr-x 10 jhuang jhuang 4,0K Jun 18 2024 Data_Tintelnot_16S
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Wuenee_Plots
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Yang_Poster
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 Data_jupnote
drwxr-xr-x 21 jhuang jhuang 4,0K Jun 18 2024 Data_parainfluenza
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 Data_snakemake_recipe
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_temp
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 Data_viGEN
drwxr-xr-x 19 jhuang jhuang 4,0K Jun 18 2024 Genomic_Data_Science
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Learn_UGENE
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 MMcPaper
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Manuscript_Epigenetics_Macrophage_Yersinia
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 Manuscript_RNAHiSwitch
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 MeDIP_Emilia_copy_DEL
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Method_biopython
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 NGS
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 Okazaki-Seq_Processing
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 RNA-NGS_Analysis_modul3_NanoStringNorm
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 RNAConSLOptV1.2
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 RNAHeliCes
drwxr-xr-x 11 jhuang jhuang 4,0K Jun 18 2024 RNA_li_HeliCes
drwxr-xr-x 10 jhuang jhuang 4,0K Jun 18 2024 RNAliHeliCes
drwxr-xr-x 10 jhuang jhuang 4,0K Jun 18 2024 RNAliHeliCes_Relatedshapes_modified
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 R_refcard
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 R_DataCamp
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 18 2024 R_cats_package
drwxr-xr-x 9 jhuang jhuang 4,0K Jun 18 2024 R_tutorials-master
drwxr-xr-x 7 jhuang jhuang 4,0K Jun 18 2024 SnakeChunks
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 align_4l_on_FJ705359
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 align_4p_on_FJ705359
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 assembly
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 bacto
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 bam2fastq_mapping_again
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 chipster
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 damian_GUI
drwxr-xr-x 4 jhuang jhuang 4,0K Jun 18 2024 enhancer-snakemake-demo
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 hg19_gene_annotations
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 interlab_comparison_DEL
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 18 2024 my_flask
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 papers
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 pangenome-snakemake_zhaoc1
drwxr-xr-x 14 jhuang jhuang 4,0K Jun 18 2024 pyflow-epilogos
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 raw_data_rnaseq_Indra
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 test_raw_data_dnaseq
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 test_raw_data_rnaseq
drwxr-xr-x 6 jhuang jhuang 4,0K Jun 18 2024 to_Francesco
drwxr-xr-x 36 jhuang jhuang 4,0K Jun 18 2024 ukepipe
drwxr-xr-x 15 jhuang jhuang 4,0K Jun 18 2024 ukepipe_nf
drwxr-xr-x 17 jhuang jhuang 4,0K Jun 18 2024 var_www_DjangoApp_mysite2_2023-05
-rwxr-xr-x 1 jhuang jhuang 59K Jun 18 2024 roentgenpass.pdf
-rwxr-xr-x 1 jhuang jhuang 9,1M Jun 18 2024 salmon_tx2gene_GRCh38.tsv
-rwxr-xr-x 1 jhuang jhuang 4,1K Jun 18 2024 salmon_tx2gene_chrHsv1.tsv
-rwxr-xr-x 1 jhuang jhuang 8,9K Jun 18 2024 'sample IDs_Lamprecht.xlsx'
-rwxr-xr-x 1 jhuang jhuang 30M Jun 18 2024 summarySCC_PM25.rds
-rwxr-xr-x 1 jhuang jhuang 0 Jun 18 2024 untitled.py
-rwxr-xr-x 1 jhuang jhuang 11M Jun 18 2024 tutorial-rnaseq.pdf
-rwxr-xr-x 1 jhuang jhuang 1,3K Jun 18 2024 x.log
-rwxr-xr-x 1 jhuang jhuang 381M Jun 18 2024 webapp.tar.gz
-rw-rw-r-- 1 jhuang jhuang 8,4K Okt 9 2024 temp
-rw-rw-r-- 1 jhuang jhuang 2,7K Okt 9 2024 temp2
drwxr-xr-x 51 jhuang jhuang 12K Feb 17 12:23 Data_Susanne_Amplicon_haplotype_analyses_RdRp_orf1_2_re
drwxr-xr-x 6 jhuang jhuang 4,0K Feb 17 12:42 Data_Susanne_WGS_unbiased
jhuang@WS-2290C:~/DATA_D$ ls -tlrh
total 56K
lrwxrwxrwx 1 jhuang jhuang 59 Apr 11 2024 Data_Soeren_RNA-seq_2023_PUBLISHING -> /media/jhuang/Elements/Data_Soeren_RNA-seq_2023_PUBLISHING/
lrwxrwxrwx 1 jhuang jhuang 32 Apr 11 2024 Data_Ute -> /media/jhuang/Elements/Data_Ute/
lrwxrwxrwx 1 jhuang jhuang 52 Apr 23 2024 Data_Marc_RNA-seq_Sepidermidis -> /media/jhuang/Titisee/Data_Marc_RNA-seq_Sepidermidis
drwxrwxr-x 2 jhuang jhuang 4,0K Mai 2 2024 Data_Patricia_Transposon
drwxrwxr-x 2 jhuang jhuang 4,0K Mai 29 2024 Books_DA_for_Life
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 18 2024 Data_Sven
-rw-rw-r-- 1 jhuang jhuang 2,9K Jul 16 2024 Datasize_calculation_based_on_coverage.txt
drwxr-xr-x 6 jhuang jhuang 4,0K Jul 23 2024 Data_Paul_HD46_1-wt_resequencing
drwxrwxr-x 2 jhuang jhuang 4,0K Jul 26 2024 Data_Sanam_DAMIAN
drwxrwxr-x 26 jhuang jhuang 12K Jul 30 2024 Data_Tam_variant_calling
drwxrwxr-x 2 jhuang jhuang 4,0K Aug 26 2024 Data_Samira_Manuscripts
drwxrwxr-x 2 jhuang jhuang 4,0K Aug 27 2024 Data_Silvia_VoltRon_Debug
drwxrwxr-x 38 jhuang jhuang 4,0K Jun 10 2025 Data_Pietschmann_229ECoronavirus_Mutations_2024
drwxrwxr-x 23 jhuang jhuang 4,0K Jun 25 2025 Data_Pietschmann_229ECoronavirus_Mutations_2025
lrwxrwxrwx 1 jhuang jhuang 63 Nov 24 16:30 Data_Birthe_Svenja_RSV_Probe3_PUBLISHING -> /media/jhuang/Elements/Data_Birthe_Svenja_RSV_Probe3_PUBLISHING
jhuang@WS-2290C:~/DATA_E$ ls -tlrh
total 119M
drwxr-xr-x 10 jhuang jhuang 4,0K Apr 18 2019 j_huang_until_201904
drwxr-xr-x 2 jhuang jhuang 4,0K Apr 29 2019 Data_2019_April
drwxr-xr-x 2 jhuang jhuang 4,0K Mai 10 2019 Data_2019_May
drwxr-xr-x 2 jhuang jhuang 4,0K Jun 17 2019 Data_2019_June
drwxr-xr-x 2 jhuang jhuang 4,0K Jul 12 2019 Data_2019_July
drwxr-xr-x 3 jhuang jhuang 4,0K Aug 29 2019 Data_2019_August
drwxr-xr-x 3 jhuang jhuang 4,0K Sep 5 2019 Data_2019_September
drwxr-xr-x 11 jhuang jhuang 4,0K Apr 18 2023 Data_Song_RNASeq_PUBLISHED
drwxr-xr-x 7 jhuang jhuang 4,0K Okt 10 2023 Data_Laura_MP_RNASeq
drwxr-xr-x 22 jhuang jhuang 12K Nov 3 2023 Data_Nicole6_HEV_Swantje2
drwxr-xr-x 17 jhuang jhuang 4,0K Nov 13 2023 Data_Becher_Damian_Picornavirus_BovHepV
-rwxr-xr-x 1 jhuang jhuang 118M Nov 28 2023 bacteria_refseq.zip
drwxr-xr-x 3 jhuang jhuang 4,0K Nov 30 2023 bacteria_refseq
drwxr-xr-x 8 jhuang jhuang 4,0K Nov 30 2023 Data_Rotavirus
drwxr-xr-x 6 jhuang jhuang 4,0K Dez 6 2023 Data_Xiaobo_10x
drwx------ 17 jhuang jhuang 4,0K Feb 7 2025 Data_Becher_Damian_Picornavirus_BovHepV_INCOMPLETE_DEL
jhuang@WS-2290C:~/DATA_Intenso$ ls -ltrh
total 4,1G
drwxr-xr-x 15 jhuang jhuang 4,0K Mär 30 2015 HOME_FREIBURG_DEL
drwxr-xr-x 2 jhuang jhuang 4,0K Aug 12 2015 150810_M03701_0019_000000000-AFJFK
drwxr-xr-x 5 jhuang jhuang 4,0K Jan 31 2017 Data_Thaiss2_Microarray
drwxr-xr-x 9 jhuang jhuang 4,0K Apr 27 2017 VirtualBox_VMs_DEL
drwxr-xr-x 7 jhuang jhuang 4,0K Apr 27 2017 'VirtualBox VMs_DEL'
drwxr-xr-x 7 jhuang jhuang 4,0K Apr 27 2017 'VirtualBox VMs2_DEL'
drwxr-xr-x 16 jhuang jhuang 4,0K Mai 12 2017 websites
drwxr-xr-x 5 jhuang jhuang 4,0K Jun 29 2017 DATA
drwxr-xr-x 149 jhuang jhuang 36K Jun 30 2017 Data_Laura
drwxr-xr-x 149 jhuang jhuang 36K Jun 30 2017 Data_Laura_2
drwxr-xr-x 3 jhuang jhuang 4,0K Jun 30 2017 Data_Laura_3
drwxr-xr-x 7 jhuang jhuang 4,0K Jul 10 2017 galaxy_tools
drwxr-xr-x 45 jhuang jhuang 32K Jul 17 2017 Downloads2
drwxr-xr-x 3 jhuang jhuang 4,0K Jul 27 2017 Downloads
drwxr-xr-x 3 jhuang jhuang 4,0K Jul 28 2017 mom-baby_com_cn
drwxr-xr-x 3 jhuang jhuang 4,0K Aug 8 2017 'VirtualBox VMs2'
drwxr-xr-x 3 jhuang jhuang 4,0K Aug 9 2017 VirtualBox_VMs
drwxr-xr-x 3 jhuang jhuang 4,0K Aug 11 2017 CLC_Data
drwxr-xr-x 6 jhuang jhuang 12K Aug 14 2017 Work_Dir2
drwxr-xr-x 7 jhuang jhuang 4,0K Aug 15 2017 Work_Dir2_SGE
drwxr-xr-x 19 jhuang jhuang 4,0K Aug 24 2017 Data_SPANDx1_Kpneumoniae_vs_Assembly1
drwxr-xr-x 3 jhuang jhuang 4,0K Aug 24 2017 MauveOutput
drwxr-xr-x 3 jhuang jhuang 4,0K Aug 31 2017 Fastqs
drwxr-xr-x 20 jhuang jhuang 4,0K Sep 7 2017 Data_Anna3_VRE_Ausbruch
drwxr-xr-x 8 jhuang jhuang 4,0K Sep 19 2017 Work_Dir_mock_broad_mockinput
drwxr-xr-x 8 jhuang jhuang 4,0K Sep 19 2017 Work_Dir_dM_broad_mockinput
drwxr-xr-x 4 jhuang jhuang 4,0K Okt 6 2017 Data_Anna8_RNASeq_static_shake_deprecated
drwxr-xr-x 24 jhuang jhuang 4,0K Okt 9 2017 PENDRIVE_cont
drwxr-xr-x 8 jhuang jhuang 4,0K Okt 23 2017 Work_Dir_WAP_broad_mockinput
drwxr-xr-x 10 jhuang jhuang 4,0K Okt 23 2017 Work_Dir_WAC_broad_mockinput
drwxr-xr-x 11 jhuang jhuang 4,0K Okt 23 2017 Work_Dir_dP_broad_mockinput
drwxr-xr-x 52 jhuang jhuang 4,0K Nov 8 2017 Data_Nicole10_16S_interlab
drwxr-xr-x 6 jhuang jhuang 4,0K Dez 6 2017 PAPERS
drwxr-xr-x 14 jhuang jhuang 16K Dez 15 2017 TB
drwxr-xr-x 5 jhuang jhuang 4,0K Dez 19 2017 Data_Anna4_SNP
drwxr-xr-x 11 jhuang jhuang 4,0K Jan 16 2018 Data_Carolin1_16S
drwxr-xr-x 2 jhuang jhuang 4,0K Jan 22 2018 ChipSeq_Raw_Data3_171009_NB501882_0024_AHNGTYBGX3
-rw-r--r-- 1 jhuang jhuang 4,0G Jan 23 2018 m_aepfelbacher_DEL.zip
drwxr-xr-x 7 jhuang jhuang 4,0K Jan 24 2018 Data_Anna7_RNASeq_Cytoscape
drwxr-xr-x 3 jhuang jhuang 4,0K Jan 24 2018 Data_Nicole9_Hund_Katze_Mega
drwxr-xr-x 39 jhuang jhuang 20K Jan 28 2018 Data_Anna2_CO6114
drwxr-xr-x 3 jhuang jhuang 4,0K Jan 28 2018 Data_Nicole3_TH17_orig
drwxr-xr-x 27 jhuang jhuang 28K Jan 28 2018 Data_Nicole1_Tropheryma_whipplei
drwxr-xr-x 16 jhuang jhuang 4,0K Jan 30 2018 results_K27
drwxr-xr-x 2 jhuang jhuang 4,0K Feb 19 2018 'VirtualBox VMs'
drwxr-xr-x 28 jhuang jhuang 12K Feb 27 2018 Data_Anna6_RNASeq
drwxr-xr-x 17 jhuang jhuang 12K Mär 1 2018 Data_Anna1_1585_RNAseq
drwxr-xr-x 21 jhuang jhuang 4,0K Mär 7 2018 Data_Thaiss1_Microarray
drwxr-xr-x 25 jhuang jhuang 12K Mär 27 2018 Data_Nicole7_Anelloviruses_Polyomavirus
drwxr-xr-x 13 jhuang jhuang 4,0K Mai 22 2018 Data_Nina1_Nicole5_1-76
drwxr-xr-x 11 jhuang jhuang 4,0K Mai 22 2018 Data_Nina1_merged
drwxr-xr-x 32 jhuang jhuang 4,0K Jun 14 2018 Data_Nicole8_Lamprecht
drwxr-xr-x 40 jhuang jhuang 16K Jul 5 2018 Data_Anna5_SNP
drwxr-xr-x 35 jhuang jhuang 4,0K Okt 12 2018 chipseq
drwxr-xr-x 107 jhuang jhuang 76K Mai 18 2019 Downloads_DEL
drwxr-xr-x 7 jhuang jhuang 4,0K Mär 17 2020 Data_Gagliani2_enriched_16S
drwxr-xr-x 17 jhuang jhuang 4,0K Mär 17 2020 Data_Gagliani1_18S_16S
drwxr-xr-x 2 jhuang jhuang 4,0K Apr 2 2020 m_aepfelbacher
drwxr-xr-x 4 jhuang jhuang 4,0K Feb 17 12:38 Data_Susanne_WGS_3amplicons
jhuang@WS-2290C:/media/jhuang/Titisee$ ls -tlrh
total 3,5G
drwxrwxrwx 1 jhuang jhuang 0 Dez 19 2017 Data_Anna4_SNP
drwxrwxrwx 1 jhuang jhuang 4,0K Jan 24 2018 Data_Anna5_SNP_rsync_error
-rwxrwxrwx 1 jhuang jhuang 9,9K Mär 21 2018 TRASH
drwxrwxrwx 1 jhuang jhuang 20K Mär 28 2018 Data_Nicole6_HEV_4_SNP_calling_PE_DEL
drwxrwxrwx 1 jhuang jhuang 4,0K Mai 22 2018 Data_Nina1_Nicole7
drwxrwxrwx 1 jhuang jhuang 8,0K Mai 24 2018 Data_Nicole6_HEV_4_SNP_calling_SE_DEL
-rwxrwxrwx 1 jhuang jhuang 3,5G Jun 14 2018 180119_M03701_0115_000000000-BFG46.zip
drwxrwxrwx 1 jhuang jhuang 4,0K Jul 10 2018 Data_Nicole10_16S_interlab_PUBLISHED
drwxrwxrwx 1 jhuang jhuang 4,0K Jul 10 2018 Anna11_assemblies
drwxrwxrwx 1 jhuang jhuang 4,0K Jul 11 2018 Anna11_trees
drwxrwxrwx 1 jhuang jhuang 4,0K Jul 24 2018 Data_Nicole6_HEV_new_orig_fastqs
drwxrwxrwx 1 jhuang jhuang 4,0K Nov 23 2018 Data_Anna9_OXA-48_or_OXA-181
drwxrwxrwx 1 jhuang jhuang 4,0K Feb 15 2019 bengal_results_v1_2018
-rwxrwxrwx 1 jhuang jhuang 9,8M Mär 22 2019 DO.pdf
drwxrwxrwx 1 jhuang jhuang 4,0K Mai 6 2019 damian_DEL
drwxrwxrwx 1 jhuang jhuang 0 Mai 20 2019 MAGpy_db
drwxrwxrwx 1 jhuang jhuang 0 Aug 3 2019 UGENE_v1_32_data_cistrome
drwxrwxrwx 1 jhuang jhuang 4,0K Aug 3 2019 UGENE_v1_32_data_ngs_classification
drwxrwxrwx 1 jhuang jhuang 52K Okt 25 2019 Data_Nicole6_HEV_Swantje
drwxrwxrwx 1 jhuang jhuang 8,0K Okt 25 2019 Data_Nico_Gagliani
drwxrwxrwx 1 jhuang jhuang 4,0K Mär 30 2020 GAMOLA2_prototyp
drwxrwxrwx 1 jhuang jhuang 8,0K Mär 31 2020 Thomas_methylation_EPIC_DO
drwxrwxrwx 1 jhuang jhuang 8,0K Jun 15 2020 Data_Nicola_Schaltenberg
drwxrwxrwx 1 jhuang jhuang 36K Jun 25 2020 Data_Nicola_Schaltenberg_PICRUSt
drwxrwxrwx 1 jhuang jhuang 12K Jan 25 2021 HOME_FREIBURG
drwxrwxrwx 1 jhuang jhuang 4,0K Okt 13 2021 Data_Francesco_16S
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 14 2022 3rd_party
drwxrwxrwx 1 jhuang jhuang 4,0K Jul 29 2022 ConsPred_prokaryotic_genome_annotation
drwxrwxrwx 1 jhuang jhuang 4,0K Aug 2 2022 'System Volume Information'
drwxrwxrwx 1 jhuang jhuang 0 Sep 16 2022 damian_v201016
drwxrwxrwx 1 jhuang jhuang 36K Jan 12 2023 Data_Holger_VRE
drwxrwxrwx 1 jhuang jhuang 32K Feb 1 2023 Data_Holger_Pseudomonas_aeruginosa_SNP
drwxrwxrwx 1 jhuang jhuang 4,0K Sep 5 2023 Eigene_Ordner_HR
drwxrwxrwx 1 jhuang jhuang 24K Sep 6 2023 GAMOLA2
drwxrwxrwx 1 jhuang jhuang 24K Sep 27 2023 Data_Anastasia_RNASeq
drwxrwxrwx 1 jhuang jhuang 24K Okt 20 2023 Data_Amir_PUBLISHED
drwxrwxrwx 1 jhuang jhuang 44K Apr 25 2024 Data_Marc_RNA-seq_Sepidermidis
drwxrwxrwx 1 jhuang jhuang 4,0K Sep 23 2024 '$RECYCLE.BIN'
drwxrwxrwx 1 jhuang jhuang 4,0K Sep 23 2024 Data_Xiaobo_10x_3
drwxrwxrwx 1 jhuang jhuang 24K Nov 28 2024 Data_Tam_DNAseq_2023_Comparative_ATCC19606_AYE_ATCC17978
drwxrwxrwx 1 jhuang jhuang 48K Dez 19 2024 Data_Holger_S.epidermidis_short
-rwxrwxrwx 1 jhuang jhuang 31 Feb 4 2025 TEMP
drwxrwxrwx 1 jhuang jhuang 12K Aug 22 11:44 Data_Holger_S.epidermidis_long
jhuang@WS-2290C:/media/jhuang/Elements(Denise_ChIPseq)$ ls -tlrh
total 11M
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 7 2019 Data_Denise_LTtrunc_H3K27me3_2_results_DEL
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 7 2019 Data_Denise_LTtrunc_H3K4me3_2_results_DEL
drwxr-xr-x 1 jhuang jhuang 28K Aug 26 2019 Data_Anna12_HAPDICS_final_not_finished_DEL
drwxr-xr-x 1 jhuang jhuang 4,0K Okt 24 2019 m_aepfelbacher_DEL
drwxr-xr-x 1 jhuang jhuang 20K Jan 14 2020 Data_Damian
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 25 2020 ST772_DEL
drwxr-xr-x 1 jhuang jhuang 160K Jan 25 2020 ALL_trimmed_part_DEL
drwxr-xr-x 1 jhuang jhuang 0 Mär 30 2020 Data_Denise_ChIPSeq_Protocol1
drwxr-xr-x 1 jhuang jhuang 44K Mai 19 2020 Data_Pietschmann_HCV_Amplicon
drwxr-xr-x 1 jhuang jhuang 60K Jun 26 2020 Data_Nicole6_HEV_ownMethod_new
-rwxr-xr-x 1 jhuang jhuang 2,5M Aug 5 2020 HD04-1.fasta
drwxr-xr-x 1 jhuang jhuang 4,0K Mai 31 2021 RNAHiSwitch_
drwxr-xr-x 1 jhuang jhuang 4,0K Mai 31 2021 RNAHiSwitch__
drwxr-xr-x 1 jhuang jhuang 8,0K Jun 17 2021 RNAHiSwitch___
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 25 2021 RNAHiSwitch_paper_
drwxr-xr-x 1 jhuang jhuang 0 Jul 7 2021 RNAHiSwitch_milestone1_DELETED
-rwxr-xr-x 1 jhuang jhuang 7,2M Jul 7 2021 RNAHiSwitch_paper.tar.gz
drwxr-xr-x 1 jhuang jhuang 4,0K Jul 12 2021 RNAHiSwitch_paper_DELETED
drwxr-xr-x 1 jhuang jhuang 12K Jul 12 2021 RNAHiSwitch_milestone1
drwxr-xr-x 1 jhuang jhuang 4,0K Aug 23 2021 RNAHiSwitch_paper
drwxr-xr-x 1 jhuang jhuang 4,0K Sep 24 2021 Ute_RNASeq_results
drwxr-xr-x 1 jhuang jhuang 4,0K Sep 24 2021 Ute_miRNA_results_38
drwxr-xr-x 1 jhuang jhuang 88K Okt 27 2021 RNAHiSwitch
drwxr-xr-x 1 jhuang jhuang 48K Mär 31 2022 Data_HepE_Freiburg_PUBLISHED
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 1 2022 Data_INTENSO_2022-06
drwxr-xr-x 1 jhuang jhuang 0 Sep 14 2022 '$RECYCLE.BIN'
drwxr-xr-x 1 jhuang jhuang 4,0K Sep 14 2022 'System Volume Information'
drwxr-xr-x 1 jhuang jhuang 4,0K Dez 7 2022 Data_Anna_Mixta_hanseatica_PUBLISHED
-rwxr-xr-x 1 jhuang jhuang 33K Dez 9 2022 coi_disclosure.docx
drwxr-xr-x 1 jhuang jhuang 20K Feb 8 2023 Data_Jingang
drwxr-xr-x 1 jhuang jhuang 4,0K Mai 30 2023 Data_Arck_16S_MMc_PUBLISHED
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 5 2023 Data_Laura_ChIPseq_GSE120945
drwxr-xr-x 1 jhuang jhuang 80K Jun 5 2023 Data_Nicole6_HEV_ownMethod
drwxr-xr-x 1 jhuang jhuang 8,0K Jul 5 2023 Data_Susanne_16S_re_UNPUBLISHED *
drwxr-xr-x 1 jhuang jhuang 4,0K Okt 12 2023 Data_Denise_ChIPSeq_Protocol2
drwxr-xr-x 1 jhuang jhuang 4,0K Okt 20 2023 Data_Caroline_RNAseq_wt_timecourse
drwxr-xr-x 1 jhuang jhuang 4,0K Okt 20 2023 Data_Caroline_RNAseq_brain_organoids
drwxr-xr-x 1 jhuang jhuang 20K Okt 20 2023 Data_Amir_PUBLISHED_DEL
drwxr-xr-x 1 jhuang jhuang 4,0K Nov 24 2023 Data_download_virus_fam
drwxr-xr-x 1 jhuang jhuang 12K Feb 22 2024 Data_Gunnar_Yersiniomics_COPYFAILED_DEL
drwxr-xr-x 1 jhuang jhuang 20K Feb 27 2024 Data_Paul_and_Marc_Epidome_batch3
-rwxr-xr-x 1 jhuang jhuang 3,0K Okt 30 2024 ifconfig_hamm.txt
drwxr-xr-x 1 jhuang jhuang 8,0K Apr 8 2025 Data_Soeren_2023_PUBLISHING
drwxr-xr-x 1 jhuang jhuang 28K Nov 24 13:34 Data_Birthe_Svenja_RSV_Probe3_PUBLISHING
drwxr-xr-x 1 jhuang jhuang 20K Jan 13 17:46 Data_Ute
drwxr-xr-x 1 jhuang jhuang 12K Feb 17 12:48 Data_Susanne_16S_UNPUBLISHED *
jhuang@WS-2290C:/media/jhuang/Seagate Expansion Drive(HOffice)$ ls -tlrh
total 19M
-rwxrwxrwx 1 jhuang jhuang 550K Jan 8 2015 SeagateExpansion.ico
-rwxrwxrwx 1 jhuang jhuang 38 Mär 27 2015 Autorun.inf
-rwxrwxrwx 2 jhuang jhuang 18M Mai 4 2017 Start_Here_Win.exe
-rwxrwxrwx 1 jhuang jhuang 1,1M Jul 7 2017 Warranty.pdf
drwxrwxrwx 1 jhuang jhuang 0 Jan 9 2018 Start_Here_Mac.app
drwxrwxrwx 1 jhuang jhuang 0 Jan 9 2018 Seagate
drwxrwxrwx 1 jhuang jhuang 0 Jun 5 2024 HomeOffice_DIR (Data_Anna_HAPDICS_RNASeq, From_Samsung_T5)
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 17 2024 DATA_COPY_FROM_178528 (copy_and_clean.sh, logfile_jhuang.log, jhuang)
drwxrwxrwx 1 jhuang jhuang 0 Sep 9 10:41 'System Volume Information'
drwxrwxrwx 1 jhuang jhuang 0 Sep 9 10:41 '$RECYCLE.BIN'
jhuang@WS-2290C:/media/jhuang/Elements(Anna_C.arnes)$ ls -trlh
total 236K
drwxrwxrwx 1 jhuang jhuang 8,0K Nov 14 2018 Data_Swantje_HEV_using_viral-ngs
drwxrwxrwx 1 jhuang jhuang 0 Dez 4 2018 VIPER_static_DEL
drwxrwxrwx 1 jhuang jhuang 4,0K Apr 4 2019 Data_Nicole6_HEV_Swantje1_blood
drwxrwxrwx 1 jhuang jhuang 24K Apr 5 2019 Data_Nicole6_HEV_benchmark
drwxrwxrwx 1 jhuang jhuang 20K Mär 12 2020 Data_Denise_RNASeq_GSE79958
drwxrwxrwx 1 jhuang jhuang 8,0K Jan 11 2022 Data_16S_Leonie_from_Nico_Gaglianis
drwxrwxrwx 1 jhuang jhuang 8,0K Jul 29 2022 Fastqs_19-21
drwxrwxrwx 1 jhuang jhuang 4,0K Aug 2 2022 'System Volume Information'
drwxrwxrwx 1 jhuang jhuang 8,0K Sep 23 2022 Data_Luise_Epidome_test
drwxrwxrwx 1 jhuang jhuang 48K Sep 27 2023 Data_Anna_C.acnes_PUBLISHED
drwxrwxrwx 1 jhuang jhuang 24K Dez 6 2023 Data_Denise_LT_DNA_Bindung
drwxrwxrwx 1 jhuang jhuang 4,0K Jan 9 2024 Data_Denise_LT_K331A_RNASeq
drwxrwxrwx 1 jhuang jhuang 12K Jan 10 2024 Data_Luise_Epidome_batch1
drwxrwxrwx 1 jhuang jhuang 28K Feb 26 2024 Data_Luise_Pseudomonas_aeruginosa_PUBLISHED
drwxrwxrwx 1 jhuang jhuang 28K Feb 27 2024 Data_Luise_Epidome_batch2
drwxrwxrwx 1 jhuang jhuang 4,0K Sep 5 2024 picrust2_out_2024_2
drwxrwxrwx 1 jhuang jhuang 4,0K Mär 11 2025 '$RECYCLE.BIN'
jhuang@WS-2290C:/media/jhuang/Seagate Expansion Drive(DATA_COPY_FROM_hamburg)$ ls -tlrh
total 19M
-rwxrwxrwx 1 jhuang jhuang 33 Feb 21 2018 Autorun.inf
-rwxrwxrwx 2 jhuang jhuang 18M Jun 21 2019 Start_Here_Win.exe
-rwxrwxrwx 1 jhuang jhuang 1,6M Jul 6 2020 Warranty.pdf
drwxrwxrwx 1 jhuang jhuang 0 Mär 10 2021 Start_Here_Mac.app
drwxrwxrwx 1 jhuang jhuang 0 Mär 10 2021 Seagate
drwxrwxrwx 1 jhuang jhuang 12K Jun 29 2022 DATA_COPY_TRANSFER_INCOMPLETE_DEL
drwxrwxrwx 1 jhuang jhuang 4,0K Dez 16 2024 DATA_COPY_FROM_hamburg
jhuang@WS-2290C:/media/jhuang/Seagate Expansion Drive(Seagate_1)$ ls -trlh
total 104G
drwxr-xr-x 1 jhuang jhuang 4,0K Okt 3 2013 RNA_seq_analysis_tools_2013
drwxr-xr-x 1 jhuang jhuang 0 Feb 28 2018 Data_Laura0
drwxr-xr-x 1 jhuang jhuang 8,0K Sep 6 2018 Data_Petra_Arck
drwxr-xr-x 1 jhuang jhuang 4,0K Sep 14 2018 Data_Martin_mycoplasma
drwxr-xr-x 1 jhuang jhuang 8,0K Dez 5 2018 chromhmm-enhancers
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 15 2019 ChromHMM_Dir
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 18 2019 Data_Denise_sT_H3K4me3
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 18 2019 Data_Denise_sT_H3K27me3
drwxr-xr-x 1 jhuang jhuang 0 Feb 13 2019 Start_Here_Mac.app
drwxr-xr-x 1 jhuang jhuang 0 Feb 13 2019 Seagate
drwxr-xr-x 1 jhuang jhuang 4,0K Feb 19 2019 Data_Nicole16_parapoxvirus
-rwxr-xr-x 1 jhuang jhuang 39G Aug 20 2019 Project_h_rohde_Susanne_WGS_unbiased_DEL.zip
drwxr-xr-x 1 jhuang jhuang 4,0K Nov 11 2019 Data_Denise_ChIPSeq_Protocol1
drwxr-xr-x 1 jhuang jhuang 8,0K Nov 13 2019 Data_ENNGS_pathogen_detection_pipeline_comparison
drwxr-xr-x 1 jhuang jhuang 4,0K Feb 18 2020 j_huang_201904_202002
-rwxr-xr-x 1 jhuang jhuang 112 Mär 2 2020 Data_Laura_ChIPseq_GSE120945
drwxr-xr-x 1 jhuang jhuang 8,0K Mär 26 2020 batch_200314_incomplete
-rwxr-xr-x 1 jhuang jhuang 65G Mär 26 2020 m_aepfelbacher.zip
drwxr-xr-x 1 jhuang jhuang 0 Mär 26 2020 m_error_DEL
drwxr-xr-x 1 jhuang jhuang 4,0K Mär 28 2020 batch_200325
drwxr-xr-x 1 jhuang jhuang 4,0K Mär 28 2020 batch_200319
drwxr-xr-x 1 jhuang jhuang 4,0K Mär 30 2020 GAMOLA2_prototyp
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 22 2020 Data_Nicola_Gagliani
drwxr-xr-x 1 jhuang jhuang 4,0K Sep 3 2020 2017-18_raw_data
drwxr-xr-x 1 jhuang jhuang 1,2M Sep 11 2020 Data_Arck_MeDIP
drwxr-xr-x 1 jhuang jhuang 4,0K Okt 16 2020 trimmed
drwxr-xr-x 1 jhuang jhuang 4,0K Dez 23 2020 Data_Nicole_16S_Christmas_2020_2
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 14 2021 j_huang_202007_202012
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 15 2021 Data_Nicole_16S_Christmas_2020
drwxr-xr-x 1 jhuang jhuang 184K Jan 18 2021 Downloads_2021-01-18_DEL
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 28 2021 Data_Laura_plasmid
drwxr-xr-x 1 jhuang jhuang 4,0K Mär 18 2021 Data_Laura_16S_2_re
drwxr-xr-x 1 jhuang jhuang 8,0K Mär 22 2021 Data_Laura_16S_2
drwxr-xr-x 1 jhuang jhuang 4,0K Mär 22 2021 Data_Laura_16S_2_re_
drwxr-xr-x 1 jhuang jhuang 8,0K Mär 23 2021 Data_Laura_16S_merged
drwxr-xr-x 1 jhuang jhuang 32K Nov 7 2022 Downloads_DEL
drwxr-xr-x 1 jhuang jhuang 12K Nov 7 2022 Data_Laura_16S
drwxr-xr-x 1 jhuang jhuang 76K Nov 9 2023 Data_Anna12_HAPDICS_final
drwxr-xr-x 1 jhuang jhuang 0 Dez 4 2023 '$RECYCLE.BIN'
drwxr-xr-x 1 jhuang jhuang 4,0K Dez 4 2023 'System Volume Information'
jhuang@WS-2290C:/media/jhuang/Seagate Expansion Drive(Seagate_2)$ ls -trlh
total 70G
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 5 2017 Data_Nicole4_TH17
-rwxr-xr-x 1 jhuang jhuang 18M Feb 9 2018 Start_Here_Win.exe
-rwxr-xr-x 1 jhuang jhuang 33 Feb 21 2018 Autorun.inf
-rwxr-xr-x 1 jhuang jhuang 1,2M Jul 26 2018 Warranty.pdf
drwxr-xr-x 1 jhuang jhuang 0 Feb 13 2019 Start_Here_Mac.app
drwxr-xr-x 1 jhuang jhuang 0 Feb 13 2019 Seagate
drwxr-xr-x 1 jhuang jhuang 4,0K Dez 20 2019 Data_Denise_RNASeq_trimmed_DEL
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 25 2020 HD12
drwxr-xr-x 1 jhuang jhuang 4,0K Jan 25 2020 Qi_panGenome
drwxr-xr-x 1 jhuang jhuang 44K Jan 25 2020 ALL
drwxr-xr-x 1 jhuang jhuang 0 Feb 14 2020 fastq_HPI_bw_2019_08_and_2020_02
-rwxr-xr-x 1 jhuang jhuang 19K Mär 12 2020 f1_R1_link.sh
-rwxr-xr-x 1 jhuang jhuang 19K Mär 12 2020 f1_R2_link.sh
drwxr-xr-x 1 jhuang jhuang 28K Mär 19 2020 rtpd_files
-rwxr-xr-x 1 jhuang jhuang 65G Apr 2 2020 m_aepfelbacher.zip
drwxr-xr-x 1 jhuang jhuang 4,0K Apr 20 2020 Data_Nicole_16S_Hamburg_Odense_Cornell_Muenster
drwxr-xr-x 1 jhuang jhuang 8,0K Apr 21 2020 HyAsP_incomplete_genomes
drwxr-xr-x 1 jhuang jhuang 4,0K Apr 25 2020 HyAsP_normal_sampled_input
drwxr-xr-x 1 jhuang jhuang 8,0K Apr 28 2020 HyAsP_complete_genomes
-rwxr-xr-x 1 jhuang jhuang 176M Mai 8 2020 video.zip
-rwxr-xr-x 1 jhuang jhuang 6,9K Jun 2 2020 sam2bedgff.pl
-rwxr-xr-x 1 jhuang jhuang 5,5K Jul 7 2020 HD04.infection.hS_vs_HD04.nose.hS_annotated_degenes.xls
drwxr-xr-x 1 jhuang jhuang 44K Jul 9 2020 ALL83
drwxr-xr-x 1 jhuang jhuang 20K Jul 9 2020 Data_Pietschmann_RSV_Probe_PUBLISHED
drwxr-xr-x 1 jhuang jhuang 8,0K Jul 27 2020 HyAsP_normal
drwxr-xr-x 1 jhuang jhuang 4,0K Jul 28 2020 Data_Manthey_16S
drwxr-xr-x 1 jhuang jhuang 8,0K Jul 29 2020 rtpd_files_DEL
drwxr-xr-x 1 jhuang jhuang 20K Aug 11 2020 HyAsP_bold
drwxr-xr-x 1 jhuang jhuang 44K Aug 17 2020 Data_HEV
drwxr-xr-x 1 jhuang jhuang 4,0K Sep 29 2020 Seq_VRE_hybridassembly
drwxr-xr-x 1 jhuang jhuang 12K Nov 11 2020 Data_Anna12_HAPDICS_raw_data_shovill_prokka
drwxr-xr-x 1 jhuang jhuang 12K Aug 10 2021 Data_Anna_HAPDICS_WGS_ALL
drwxr-xr-x 1 jhuang jhuang 20K Aug 10 2021 Data_HEV_Freiburg_2020
drwxr-xr-x 1 jhuang jhuang 20K Okt 27 2021 Data_Nicole_HDV_Recombination_PUBLISHED
-rwxr-xr-x 1 jhuang jhuang 905K Feb 8 2022 s_hero2x
-rwxr-xr-x 1 jhuang jhuang 5,5G Feb 25 2022 201030_M03701_0207_000000000-J57B4.zip
-rwxr-xr-x 1 jhuang jhuang 4,9K Mär 21 2022 README
-rwxr-xr-x 1 jhuang jhuang 4,9K Mär 21 2022 'README(1)'
-rwxr-xr-x 1 jhuang jhuang 848 Mär 28 2022 dna2.fasta.fai
-rwxr-xr-x 1 jhuang jhuang 17K Mär 28 2022 91.pep
-rwxr-xr-x 1 jhuang jhuang 9,1K Mär 28 2022 91.orf
-rwxr-xr-x 1 jhuang jhuang 222 Mär 28 2022 91.orf.fai
-rwxr-xr-x 1 jhuang jhuang 1,1M Mär 31 2022 dgaston-dec-06-2012-121211124858-phpapp01.pdf
-rwxr-xr-x 1 jhuang jhuang 5,2K Apr 4 2022 tileshop.fcgi
-rwxr-xr-x 1 jhuang jhuang 765K Apr 4 2022 ppat.1009304.s016.tif
-rwxr-xr-x 1 jhuang jhuang 4,1K Mai 2 2022 sequence.txt
-rwxr-xr-x 1 jhuang jhuang 4,0K Mai 2 2022 'sequence(1).txt'
-rwxr-xr-x 1 jhuang jhuang 3,7K Mai 23 2022 GSE128169_series_matrix.txt.gz
-rwxr-xr-x 1 jhuang jhuang 4,0K Mai 23 2022 GSE128169_family.soft.gz
drwxr-xr-x 1 jhuang jhuang 40K Mär 20 2023 Data_Anna_HAPDICS_RNASeq
drwxr-xr-x 1 jhuang jhuang 1,3M Apr 4 2023 Data_Christopher_MeDIP_MMc_PUBLISHED
drwxr-xr-x 1 jhuang jhuang 8,0K Jun 28 2023 Data_Gunnar_Yersiniomics_IMCOMPLETE_DEL
drwxr-xr-x 1 jhuang jhuang 28K Feb 12 2024 Data_Denise_RNASeq
drwxr-xr-x 1 jhuang jhuang 4,0K Apr 5 2024 'System Volume Information'
drwxr-xr-x 1 jhuang jhuang 0 Apr 5 2024 '$RECYCLE.BIN'
jhuang@WS-2290C:/media/jhuang/Elements(An14_RNAs)$ ls -tlrh
total 284K
drwxr-xr-x 1 jhuang jhuang 8,0K Aug 7 2017 Data_Anna10_RP62A
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 15 2018 Data_Nicole12_16S_Kluwe_Bunders
drwxr-xr-x 1 jhuang jhuang 4,0K Nov 30 2018 chromhmm-enhancers
drwxr-xr-x 1 jhuang jhuang 0 Apr 1 2019 Data_Denise_sT_Methylation
drwxr-xr-x 1 jhuang jhuang 0 Apr 1 2019 Data_Denise_LTtrunc_Methylation
drwxr-xr-x 1 jhuang jhuang 12K Apr 29 2019 Data_16S_arckNov
drwxr-xr-x 1 jhuang jhuang 4,0K Mai 29 2019 Data_Tabea_RNASeq
-rwxr-xr-x 1 jhuang jhuang 4,6K Mai 29 2019 nr_gz_README
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 5 2019 j_huang_raw_fq
drwxr-xr-x 1 jhuang jhuang 0 Jun 7 2019 'System Volume Information'
drwxr-xr-x 1 jhuang jhuang 0 Jun 7 2019 '$RECYCLE.BIN'
drwxr-xr-x 1 jhuang jhuang 36K Jun 18 2019 host_refs
drwxr-xr-x 1 jhuang jhuang 0 Jun 18 2019 Vraw
drwxr-xr-x 1 jhuang jhuang 68K Jul 29 2019 Data_Susanne_Amplicon_RdRp_orf1_2 *
drwxr-xr-x 1 jhuang jhuang 4,0K Aug 6 2019 tmp
drwxr-xr-x 1 jhuang jhuang 28K Sep 4 2020 Data_RNA188_Paul_Becher
drwxr-xr-x 1 jhuang jhuang 4,0K Nov 3 2020 Data_ChIPSeq_Laura
drwxr-xr-x 1 jhuang jhuang 12K Mai 7 2021 Data_16S_arckNov_review_PUBLISHED
drwxr-xr-x 1 jhuang jhuang 8,0K Mai 7 2021 Data_16S_arckNov_re
drwxr-xr-x 1 jhuang jhuang 20K Mai 25 2021 Fastqs
drwxr-xr-x 1 jhuang jhuang 4,0K Aug 9 2021 Data_Tabea_RNASeq_submission
drwxr-xr-x 1 jhuang jhuang 4,0K Aug 27 2021 Data_Anna_Cutibacterium_acnes_DEL
drwxr-xr-x 1 jhuang jhuang 0 Sep 16 2021 Data_Silvia_RNASeq_SUBMISSION
drwxr-xr-x 1 jhuang jhuang 4,0K Feb 9 2022 Data_Hannes_ChIPSeq
drwxr-xr-x 1 jhuang jhuang 4,0K Jul 5 2022 Data_Anna14_RNASeq_to_be_DEL
drwxr-xr-x 1 jhuang jhuang 40K Dez 15 2022 Data_Pietschmann_RSV_Probe2_PUBLISHED
drwxr-xr-x 1 jhuang jhuang 0 Dez 16 2022 Data_Holger_Klebsiella_pneumoniae_SNP_PUBLISHING
drwxr-xr-x 1 jhuang jhuang 4,0K Jun 29 2023 Data_Anna14_RNASeq_plus_public
jhuang@WS-2290C:/media/jhuang/Elements(Indra_HAPDICS)$ ls -trlh
total 452K
drwxrwxrwx 1 jhuang jhuang 20K Jul 3 2018 Data_Anna11_Sepdermidis_DEL
drwxrwxrwx 1 jhuang jhuang 20K Jul 12 2018 HD15_without_10
drwxrwxrwx 1 jhuang jhuang 12K Jul 12 2018 HD31
drwxrwxrwx 1 jhuang jhuang 20K Jul 12 2018 HD33
drwxrwxrwx 1 jhuang jhuang 20K Jul 12 2018 HD39
drwxrwxrwx 1 jhuang jhuang 20K Jul 12 2018 HD43
drwxrwxrwx 1 jhuang jhuang 20K Jul 12 2018 HD46
drwxrwxrwx 1 jhuang jhuang 20K Jul 12 2018 HD15_with_10
drwxrwxrwx 1 jhuang jhuang 12K Jul 13 2018 HD26
drwxrwxrwx 1 jhuang jhuang 20K Jul 13 2018 HD59
drwxrwxrwx 1 jhuang jhuang 12K Jul 13 2018 HD25
drwxrwxrwx 1 jhuang jhuang 20K Jul 16 2018 HD21
drwxrwxrwx 1 jhuang jhuang 20K Jul 17 2018 HD17
drwxrwxrwx 1 jhuang jhuang 24K Sep 24 2018 HD04
drwxrwxrwx 1 jhuang jhuang 20K Mär 5 2019 Data_Anna11_Pair1-6_P6
drwxrwxrwx 1 jhuang jhuang 4,0K Aug 15 2019 Data_Anna12_HAPDICS_HyAsP
drwxrwxrwx 1 jhuang jhuang 68K Dez 27 2019 HAPDICS_hyasp_plasmids
drwxrwxrwx 1 jhuang jhuang 8,0K Jan 14 2021 Data_Anna_HAPDICS_review
-rwxrwxrwx 1 jhuang jhuang 9,6K Jan 26 2021 data_overview.txt
drwxrwxrwx 1 jhuang jhuang 4,0K Jan 29 2021 align_assem_res_DEL
drwxrwxrwx 1 jhuang jhuang 0 Jun 8 2021 'System Volume Information'
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 8 2021 EXCHANGE_DEL
drwxrwxrwx 1 jhuang jhuang 8,0K Aug 30 2021 Data_Indra_H3K4me3_public
drwxrwxrwx 1 jhuang jhuang 4,0K Feb 17 2022 Data_Gunnar_MS
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 2 2022 '$RECYCLE.BIN'
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 2 2022 UKE_DELLWorkstation_C_Users_indbe_Desktop
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 2 2022 Linux_DELLWorkstation_C_Users_indbe_VirtualBoxVMs
drwxrwxrwx 1 jhuang jhuang 4,0K Jun 23 2022 Data_Anna_HAPDICS_RNASeq_rawdata
drwxrwxrwx 1 jhuang jhuang 8,0K Jun 23 2022 Data_Indra_H3K27ac_public
drwxrwxrwx 1 jhuang jhuang 28K Feb 22 2023 Data_Holger_Klebsiella_pneumoniae_SNP_PUBLISHING
drwxrwxrwx 1 jhuang jhuang 4,0K Dez 9 2024 DATA_INDRA_RNASEQ
drwxrwxrwx 1 jhuang jhuang 4,0K Dez 9 2024 DATA_INDRA_CHIPSEQ
jhuang@WS-2290C:/media/jhuang/Elements(jhuang_*)$ ls -ltrh
total 5,0M
-rwxr-xr-x 1 jhuang jhuang 657K Jul 9 2021 'Install Western Digital Software for Windows.exe'
-rwxr-xr-x 1 jhuang jhuang 498K Jul 9 2021 'Install Western Digital Software for Mac.dmg'
drwxr-xr-x 2 jhuang jhuang 1,0M Mai 17 2023 'System Volume Information'
drwxr-xr-x 2 jhuang jhuang 1,0M Aug 26 2024 '$RECYCLE.BIN'
drwxr-xr-x 11 jhuang jhuang 1,0M Feb 4 2025 20250203_FS10003086_95_BTR67811-0621
jhuang@WS-2290C:/media/jhuang/Smarty$ ls -tlrh
total 140K
drwx------ 2 jhuang jhuang 16K Mär 14 2018 lost+found
drwxrwxrwx 21 jhuang jhuang 68K Jun 10 2022 Blast_db
drwxrwxr-x 2 jhuang jhuang 4,0K Sep 5 2022 temporary_files_DEL
drwxrwxr-x 9 jhuang jhuang 12K Sep 6 2022 ALIGN_ASSEM
drwxr-xr-x 19 jhuang jhuang 4,0K Sep 29 2022 Data_Paul_Staphylococcus_epidermidis
drwxrwxr-x 11 jhuang jhuang 4,0K Jan 26 2023 Data_16S_Degenhardt_Marius_DEL
drwxrwxr-x 16 jhuang jhuang 4,0K Jun 28 2023 Data_Gunnar_Yersiniomics_DEL
drwxrwxr-x 6 jhuang jhuang 4,0K Jul 5 2023 Data_Manja_RNAseq_Organoids_Virus
drwxrwxr-x 19 jhuang jhuang 12K Sep 27 2023 Data_Emilia_MeDIP
drwxr-xr-x 14 jhuang jhuang 4,0K Okt 30 2023 DjangoApp_Backup_2023-10-30
drwxrwxr-x 5 jhuang jhuang 4,0K Apr 19 2024 ref
drwxrwxr-x 4 jhuang jhuang 4,0K Jul 22 2025 Data_Michelle_RNAseq_2025_raw_data_DEL_AFTER_UPLOAD_GEO