癌症治疗方案涉及疗效和副作用之间的重要权衡。为了将患者分配到最佳治疗方案,必须能够在诊断后不久预测他们的风险水平。在临床环境中生成的分子数据的增加使得预测更加准确成为可能,但是需要采用复杂的方法来训练预测算法。在我们 另外的文章 中曾經讨论过机器学习主题,现在我们专注于使用一种称为随机森林的方法来预测癌症生存。
要点:
- 机器学习能够基于高通量数据(如基因组表达和突变数据)对患者的风险进行分层。
- 随机森林是一种强大且流行的算法,旨在提高较简单模型的泛化能力。
- 为了预测生存,使用的预测算法和性能指标必须能够考虑到被审查的数据。
- 在训练模型的同时,评估预测模型在未使用过的测试数据上的性能非常重要。
介绍
上一次,我们介绍了机器学习的关键概念,考虑了一个简单的预测治疗反应的情况。现在,我们将更详细地了解预测算法的内部工作原理,这次是在预测生存的情况下。
生存分析通常使用Kaplan-Meier估计(用于单个预测变量或输入变量)和Cox回归(用于少量预测变量)。然而,这些方法不容易适用于具有复杂的非线性预测变量之间的高维数据(即基因组测量)。机器学习正是为这种问题而设计的解决方案。
看到森林中的树
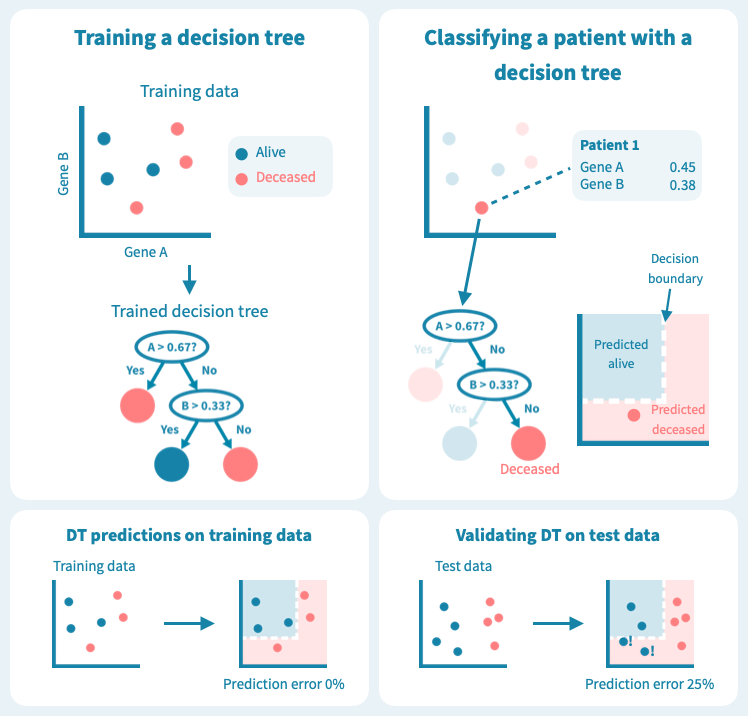
一种常见的适用于生存分析的机器学习算法称为随机森林。为了理解随机森林的工作原理,让我们首先考虑一个它所基于的更简单的算法:决策树。
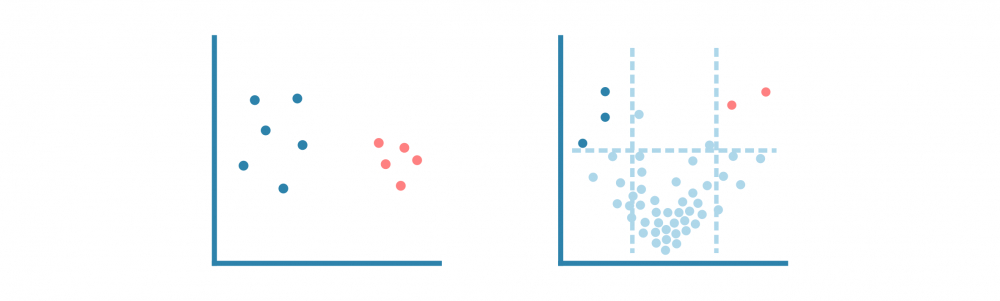
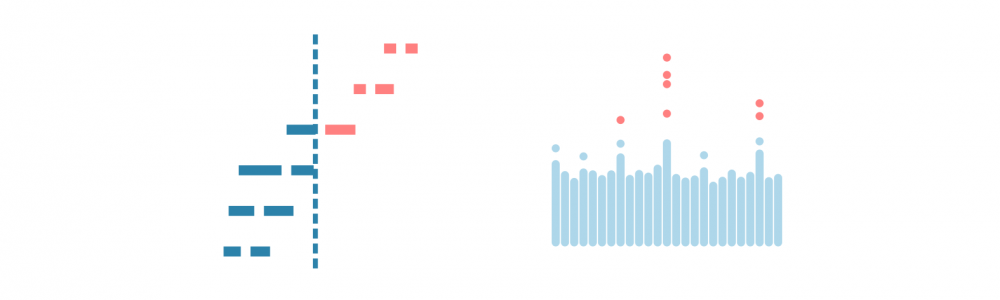
下图显示了决策树在一组六个患者(三个仍然存活,三个在诊断后5年去世)上进行训练的玩具示例。使用仅来自原发肿瘤活检的两个基因的表达水平,训练后的树能够完美地分离两组患者。然而,树在测试数据上的表现——8个新的患者,这些患者未在模型训练中使用——更差,有两个患者被错分类。这描绘了机器学习中的一个核心挑战:预测模型在训练数据上可能表现非常好,但在未见过的测试数据上表现很差。过度拟合或者训练数据集过于偏斜或太小,可能导致泛化能力差。
什么是随机森林?
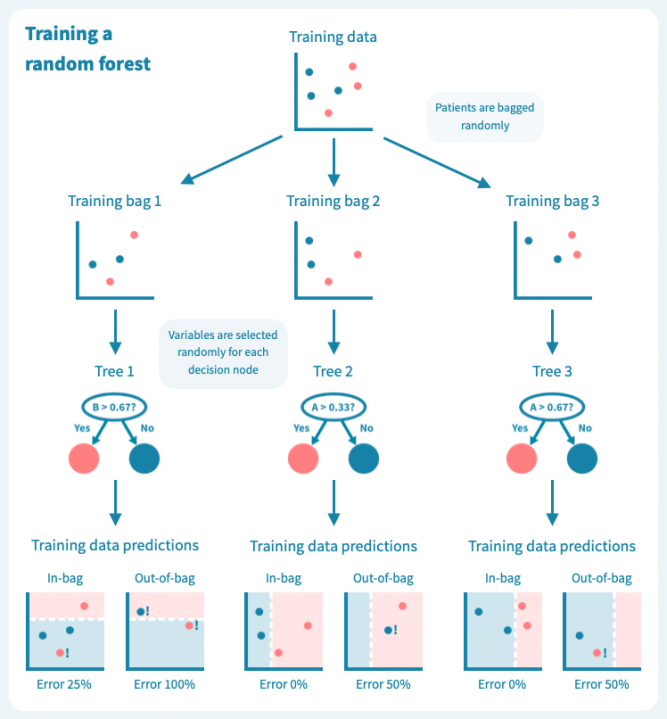
过拟合是决策树的已知问题,随机森林便应运而生,以解决这个问题。随机森林由多个决策树组成,这些树是通过对训练数据(例如患者)和预测变量(基因)进行随机子抽样而训练的。这导致了一组具有偏差但各有特点的决策树。随机森林的预测是其个别树的预测的组合(例如多数投票)。因此,随机森林是一种集成分类器,也是一种众包算法。这种方法有效地减少了个别决策树的过拟合问题。
下面的图示展示了一个随机森林(仅由三棵小树组成)在我们的玩具数据上训练的情况。每棵树都使用完整训练集(一个包)的随机样本进行训练,每棵树的每个决策节点由随机选择的基因定义。虽然树在其包中的患者表现良好(特定于树的训练数据),但它们在包外患者身上表现不佳。显然,限制在训练树时使用数据会降低其性能,这不应让我们感到惊讶!
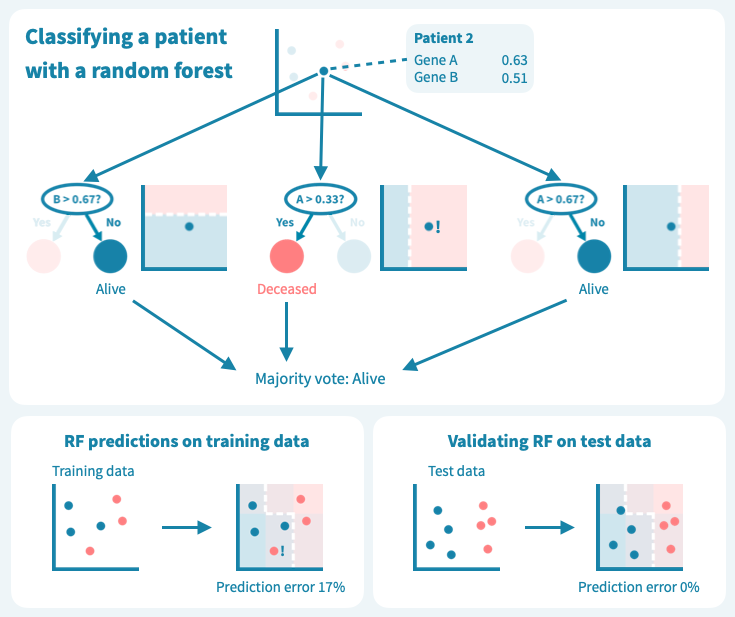
然而,这些树的组合预测在测试数据上的表现却很好——事实上,比第一个例子中的简单决策树模型要好。下面的图示展示了每个决策树如何为随机森林的集成预测做出贡献,从而在由预测变量或输入变量定义的空间中形成一个组合决策边界。
从玩具数据到真实世界数据
虽然我们上面的例子数据包含的患者和测量值比任何真实世界的预测问题所需的要少,但它使我们了解了算法如何基于输入变量(基因表达值)推导目标变量(例如生存状态)的预测,以及如何通过机器学习算法设计来应对泛化能力的挑战和如何对抗过拟合。
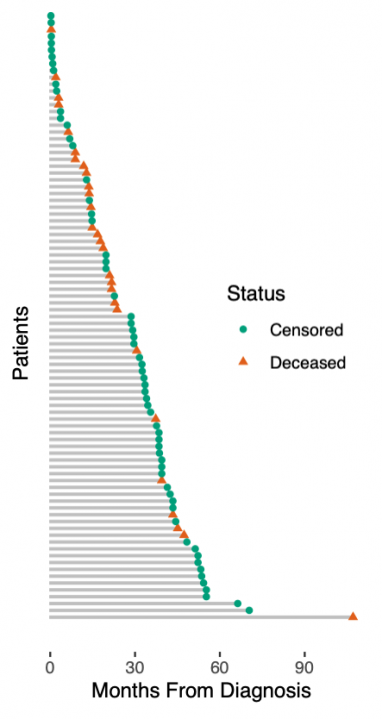
展示真实世界数据的生存预测之前,我们必须承认我们玩具数据中的另一个简化。我们假设一个简单的死亡或生存分类任务。实际上,生存数据更加复杂:患者要么死亡,要么被审查。被审查的患者按照最后一次随访仍然活着,之后他们可能很快死亡,也可能过着幸福的生活——但我们不知道。这种所谓的right-censoring是一种缺失的形式,任何生存分析都必须考虑到这一点。右图说明了生存数据:从诊断到死亡或最后一次随访的时间是已知的,而在最后一次随访时仍然存活的患者被认为是被审查的。
一种适应于生存预测的随机森林(RSF)——随机生存森林,应用于这种类型的数据。它不是将患者分类为死亡或生存,而是旨在根据其估计的风险分层患者。因此,这种模型的预测不是二元分类,而是连续的风险评分。
评估模型
由于模型输出(预测)是连续的风险评分而不是易于验证的死亡或生存类别,如何评估模型的性能?一个好的答案是c指数或一致性指数。 c指数计算为具有更高预测风险的患者对中实际死亡的比例。低预测风险的患者对不能评估一致性的患者对被删减,这样的患者对是由于审查而必须接受的限制。 c指数为0.5对应于随机的、无用的分类器,而1表示按风险对患者排序的完美结果。这种c指数的解释可能会让你想起我们在 另一篇文章 中讨论的AUC的解释。实际上,c指数是对非二元预测问题的AUC的一般化。
评估模型性能的另一种方法是按预测风险对患者进行分组,绘制组特定的Kaplan-Meier估计量,并使用log-rank检验比较组。预测模型越好,高风险组和低风险组之间的区分就越明显。重要的是要基于测试数据而不仅仅是训练数据来评估这些性能指标。前者可能会更糟糕,但比后者更有信息量。
使用随机生存森林预测肝癌生存率
下图显示了使用来自癌症基因组图谱(TCGA)的公共数据,通过对肝癌患者的原发肿瘤的基因表达谱进行分层的结果。在TCGA-LIHC数据集的369名肝癌患者中,我们随机选择了80%,即269名患者,作为训练集来构建随机生存森林。其余的20%,即73名患者,被保留为测试集。在训练数据上的风险分层表现很好,c-指数为0.925,通过中位数风险进行二元分组可以产生具有明显不同生存模式和log-rank P值低于0.0001的风险组。
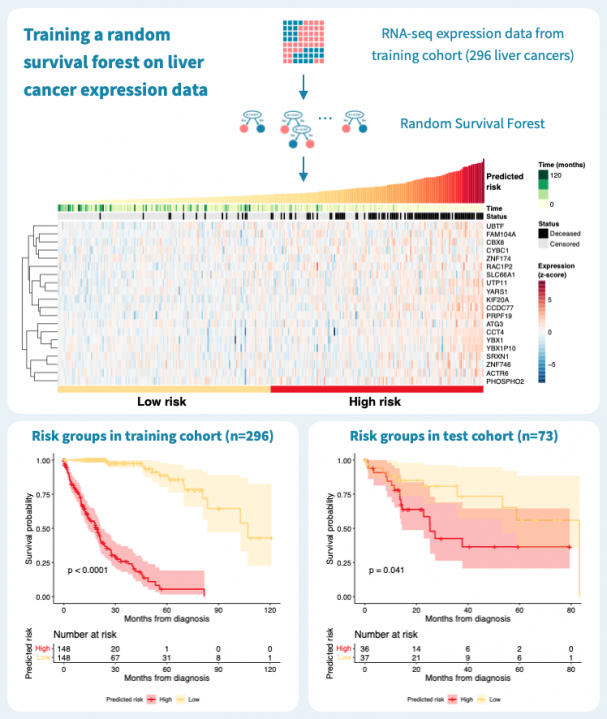
那么这个模型的推广效果如何呢?对73名患者的测试集进行的预测具有0.61的c-指数,并且二元分组的P值为0.041。性能明显优于什么都没有,但比训练数据上的性能差。这突显了使用单独的测试数据来评估模型的重要性。毕竟,训练数据上的表现对于构建模型和最终临床应用中确定的生物标志物的适用性告诉的很少。
超越基因表达
在上面的示例中,我们只考虑了基因表达水平作为预测因子或潜在的生物标志物。机器学习模型(如随机森林)的一个重要优势是,它们允许轻松地将任何可用数据(如临床变量和突变)作为输入集成到模型中。数据越多,预测就越好,只要应用的训练和验证方案确保了泛化性!
了解更多
另外 的一篇文章,介绍了机器学习的概念,并重点介绍了治疗反应预测。