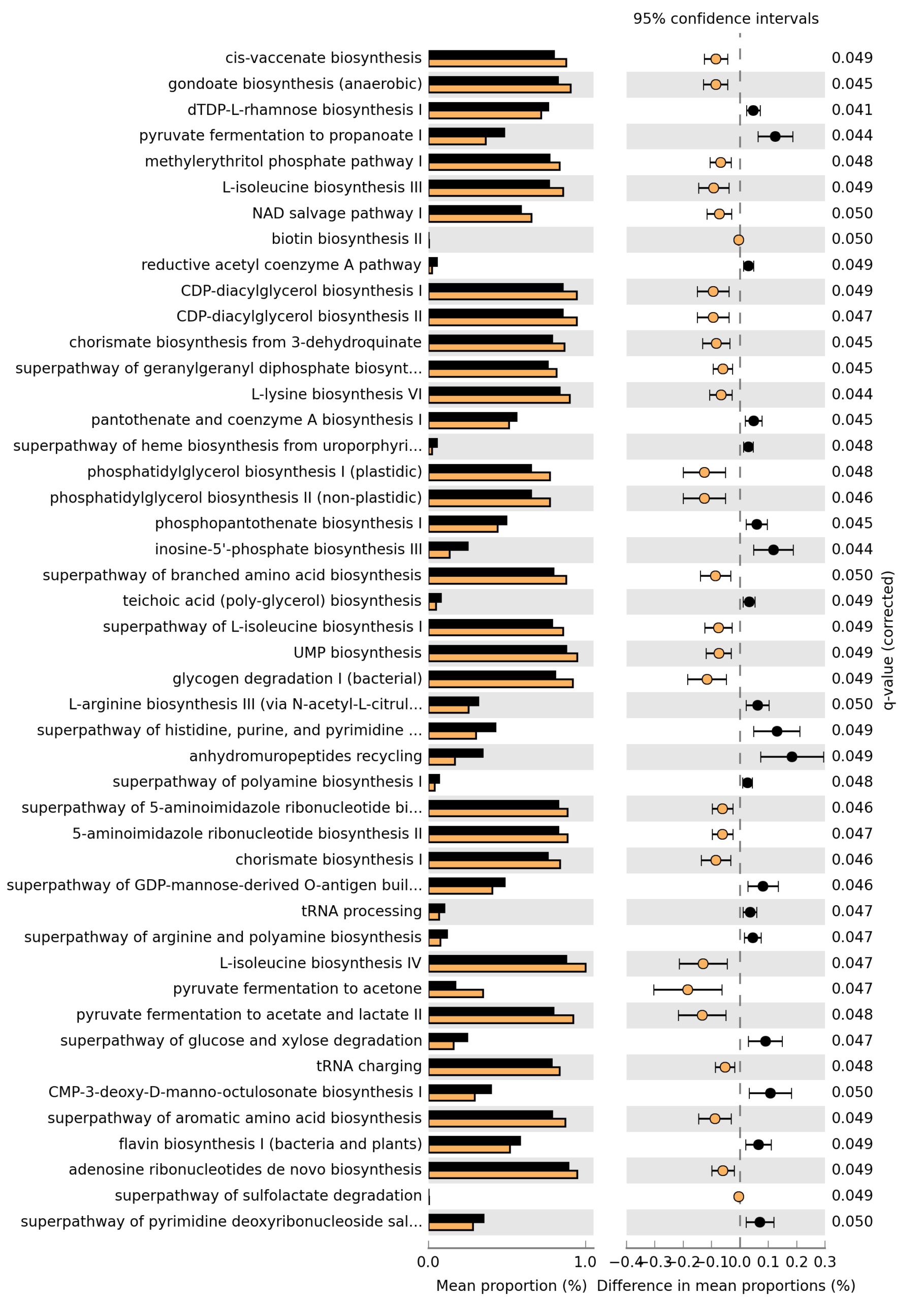
Path: ~/DATA/Data_Michelle/MS_RNAseq_Venn_2026
This post documents the complete, reproducible pipeline used to generate a 3-circle Venn diagram integrating RNA-seq, exoproteome, and MS SIP (Proximity + ALFApulldown) datasets, harmonized at the locus-tag level.
The pipeline is intentionally verbose: all transformations, mappings, and decisions are logged so the analysis can be reconstructed later.
Overview of the Three Sets
Circle 1 — RNA-seq
Union of all significantly regulated genes across six comparisons
Criteria: padj < 0.05 and |log2FC| > 2
Circle 2 — Exoproteome
Union of six exoproteome conditions (MH/TSB × 1h, 4h, 18h)
Protein IDs (UniProt / UniParc) are mapped to locus tags via BLAST
Direction is defined without a cutoff:
log2FC > 0 → UPlog2FC < 0 → DOWN
Circle 3 — MS SIP (ProALFA)
Union of four datasets:
- Proximity 4h / 18h
- ALFApulldown 4h / 18h
Protein IDs are mapped to locus tags via BLAST
Outputs
The pipeline produces:
Step A — Fetch exoproteome FASTA from UniProt
Exoproteome Excel sheets contain UniProt accessions, while RNA-seq uses locus tags.
To enable mapping, protein sequences are downloaded from UniProt and UniParc.
Script
01_fetch_uniprot_fasta_from_exoproteome_excel.py
What it does
- Reads exoproteome sheets from Excel
- Extracts UniProt / UniParc IDs
- Downloads FASTA via UniProt REST (with caching + fallback to UniParc)
-
Writes:
- one FASTA per sheet
- mapping tables
- UP / DOWN ID lists
- run log
Bash: fetch exoproteome FASTAs
python3 01_fetch_uniprot_fasta_from_exoproteome_excel.py \
--excel "Zusammenfassung SIP und Exoproteome.xlsx" \
--outdir ./exoproteome_fastas \
--cache ./uniprot_cache
Step B — Normalize FASTA headers (Exoproteome + MS)
FASTA headers from UniProt and MS are heterogeneous and may contain duplicates.
All FASTA files are normalized to canonical, unique protein IDs.
Script
02_normalize_fasta_headers.py
What it does
- Extracts canonical IDs (
sp|…|, tr|…|, raw headers)
- Enforces uniqueness (
__dupN suffix if needed)
- Writes an ID mapping table for traceability
Bash: normalize exoproteome FASTAs
mkdir -p normalized_fastas
for f in \
Exoproteome_MH_1h.fasta Exoproteome_MH_4h.fasta Exoproteome_MH_18h.fasta \
Exoproteome_TSB_1h.fasta Exoproteome_TSB_4h.fasta Exoproteome_TSB_18h.fasta
do
python3 02_normalize_fasta_headers.py \
exoproteome_fastas/"$f" \
"$f" \
"$f.idmap.tsv"
done
Bash: normalize MS (Proximity / ALFA) FASTAs
for f in \
Proximity_4h.fasta Proximity_18h.fasta \
ALFApulldown_4h.fasta ALFApulldown_18h.fasta
do
python3 02_normalize_fasta_headers.py \
../MS_GO_enrichments/"$f" \
"$f" \
"$f.idmap.tsv"
done
Step C — BLAST mapping to reference proteome
All exoproteome and MS proteins are mapped to locus tags using BLASTP against the reference proteome.
Bash: merge exoproteome FASTAs
cat Exoproteome_MH_1h.fasta Exoproteome_MH_4h.fasta Exoproteome_MH_18h.fasta \
Exoproteome_TSB_1h.fasta Exoproteome_TSB_4h.fasta Exoproteome_TSB_18h.fasta \
> Exoproteome_ALL.fasta
Bash: build BLAST database (once)
makeblastdb -in CP020463_protein.fasta \
-dbtype prot \
-out CP020463_ref_db
Bash: BLAST exoproteome
blastp -query Exoproteome_ALL.fasta \
-db CP020463_ref_db \
-out Exoproteome_ALL_vs_ref.blast.tsv \
-outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen" \
-evalue 1e-10 \
-max_target_seqs 5 \
-num_threads 16
Bash: select best hit per query (exoproteome)
awk -F'\t' 'BEGIN{OFS="\t"}{
key=$1; bits=$12+0; e=$11+0
if(!(key in best) || bits>best_bits[key] || (bits==best_bits[key] && e<best_e[key])){
best[key]=$0; best_bits[key]=bits; best_e[key]=e
}
} END{for(k in best) print best[k]}' \
Exoproteome_ALL_vs_ref.blast.tsv \
| sort -t$'\t' -k1,1 \
> Exoproteome_ALL_vs_ref.besthit.tsv
Bash: BLAST MS datasets
for base in Proximity_4h Proximity_18h ALFApulldown_4h ALFApulldown_18h; do
blastp -query "${base}.fasta" \
-db CP020463_ref_db \
-out "${base}_vs_ref.blast.tsv" \
-outfmt "6 qseqid sseqid pident length mismatch gapopen qstart qend sstart send evalue bitscore qlen slen" \
-evalue 1e-10 \
-max_target_seqs 5 \
-num_threads 16
done
Bash: best-hit selection (MS)
for base in Proximity_4h Proximity_18h ALFApulldown_4h ALFApulldown_18h; do
awk -F'\t' 'BEGIN{OFS="\t"}{
q=$1; b=$12+0; e=$11+0
if(!(q in best) || b>bb[q] || (b==bb[q] && e<be[q])){
best[q]=$0; bb[q]=b; be[q]=e
}
} END{for(q in best) print best[q]}' \
"${base}_vs_ref.blast.tsv" \
| sort -t$'\t' -k1,1 \
> "${base}_vs_ref.besthit.tsv"
done
Step D — R wrapper: integration, Venn, Excel, LOG
Script
03_wrapper_3circles_venn_RNAseq_Exo_ProALFA.R
What it does
- Reads RNA-seq Excel files (6 comparisons)
- Reads exoproteome and MS sheets
- Applies BLAST best-hit mappings
- Builds union sets and intersections
-
Generates:
- Venn plot
- detailed Excel workbook
- comprehensive log file
Bash: run the final integration
Rscript 03_wrapper_3circles_venn_RNAseq_Exo_ProALFA.R
Scripts Used (for traceability)
01_fetch_uniprot_fasta_from_exoproteome_excel.py
# see script file for full implementation
02_normalize_fasta_headers.py
# see script file for full implementation
03_wrapper_3circles_venn_RNAseq_Exo_ProALFA.R
# see script file for full implementation
Final Note
Keeping Python, Bash, and R steps together is intentional:
this pipeline is meant to be auditable, reproducible, and explainable, even months later when details are fuzzy.
If you want, I can next:
- condense this into a README.md
- split it into Methods vs Reproducibility
- or add a flowchart of the pipeline steps
Scripts Used
01_fetch_uniprot_fasta_from_exoproteome_excel.py
#!/usr/bin/env python3
"""
01_fetch_uniprot_fasta_from_exoproteome_excel.py (v3)
MS log2FC direction rule (no cutoff):
Up : log2FC > 0
Down : log2FC < 0
Zero : log2FC == 0
NA : missing/non-numeric log2FC
Reads per sheet:
- protein IDs from column A (index 0)
- log2FC from column D (index 3)
Outputs per sheet:
- {sheet}.fasta
- {sheet}.mapping.tsv
- {sheet}.up.ids.txt / {sheet}.down.ids.txt
- run.log
UniParc catching method:
1) Try UniProtKB FASTA: /uniprotkb/{ACC}.fasta
2) If missing: UniParc search -> UPI
3) Fetch UniParc FASTA: /uniparc/{UPI}.fasta
"""
import argparse
import os
import re
import time
from typing import Optional, Tuple
import pandas as pd
import requests
DEFAULT_SHEETS = [
"Exoproteome MH 1h",
"Exoproteome MH 4h",
"Exoproteome MH 18h",
"Exoproteome TSB 1h",
"Exoproteome TSB 4h",
"Exoproteome TSB 18h",
]
UNIPROTKB_FASTA = "https://rest.uniprot.org/uniprotkb/{acc}.fasta"
UNIPARC_FASTA = "https://rest.uniprot.org/uniparc/{upi}.fasta"
UNIPARC_SEARCH = "https://rest.uniprot.org/uniparc/search"
def sanitize_name(s: str) -> str:
s = s.strip()
s = re.sub(r"[^\w\-\.]+", "_", s)
s = re.sub(r"_+", "_", s)
return s
def parse_id_cell(raw: str) -> Optional[str]:
if raw is None:
return None
s = str(raw).strip()
if not s or s.lower() in {"nan", "na"}:
return None
s = s.split()[0].strip()
# header like tr|ACC|NAME
if "|" in s:
parts = s.split("|")
if len(parts) >= 2 and parts[0] in {"tr", "sp"}:
s = parts[1]
else:
s = parts[0]
if re.fullmatch(r"UPI[0-9A-F]{10,}", s):
return s
if re.fullmatch(r"[A-Z0-9]{6,10}", s):
return s
return None
def to_float(x) -> Optional[float]:
if x is None:
return None
try:
s = str(x).strip()
if s == "" or s.lower() in {"nan", "na"}:
return None
s = s.replace(",", ".")
return float(s)
except Exception:
return None
def classify_direction_no_cutoff(log2fc: Optional[float]) -> str:
if log2fc is None:
return "NA"
if log2fc > 0:
return "Up"
if log2fc < 0:
return "Down"
return "Zero"
def extract_ids_and_fc_from_sheet(excel_path: str, sheet: str) -> pd.DataFrame:
df = pd.read_excel(excel_path, sheet_name=sheet, header=None)
if df.shape[1] < 4:
raise ValueError(f"Sheet '{sheet}' has <4 columns, cannot read column D (log2FC).")
out_rows = []
for _, row in df.iterrows():
pid = parse_id_cell(row.iloc[0]) # col A
if not pid:
continue
log2fc = to_float(row.iloc[3]) # col D
out_rows.append({"protein_id": pid, "log2fc": log2fc})
out = pd.DataFrame(out_rows)
if out.empty:
return out
# de-duplicate by protein_id (keep first non-NA log2fc if possible)
out["log2fc_isna"] = out["log2fc"].isna()
out = out.sort_values(["protein_id", "log2fc_isna"]).drop_duplicates("protein_id", keep="first")
out = out.drop(columns=["log2fc_isna"]).reset_index(drop=True)
return out
def http_get_text(session: requests.Session, url: str, params: dict = None,
retries: int = 4, backoff: float = 1.5) -> Optional[str]:
for attempt in range(retries):
r = session.get(url, params=params, timeout=60)
if r.status_code == 200:
return r.text
if r.status_code in (429, 500, 502, 503, 504):
time.sleep(backoff * (attempt + 1))
continue
return None
return None
def fetch_uniprotkb_fasta(session: requests.Session, acc: str) -> Optional[str]:
return http_get_text(session, UNIPROTKB_FASTA.format(acc=acc))
def resolve_upi_via_uniparc_search(session: requests.Session, acc: str) -> Optional[str]:
params = {"query": acc, "format": "tsv", "fields": "upi", "size": 1}
txt = http_get_text(session, UNIPARC_SEARCH, params=params)
if not txt:
return None
lines = [ln.strip() for ln in txt.splitlines() if ln.strip()]
if len(lines) < 2:
return None
upi = lines[1].split("\t")[0].strip()
if re.fullmatch(r"UPI[0-9A-F]{10,}", upi):
return upi
return None
def fetch_uniparc_fasta(session: requests.Session, upi: str) -> Optional[str]:
return http_get_text(session, UNIPARC_FASTA.format(upi=upi))
def cache_get(cache_dir: str, key: str) -> Optional[str]:
if not cache_dir:
return None
path = os.path.join(cache_dir, f"{key}.fasta")
if os.path.exists(path) and os.path.getsize(path) > 0:
with open(path, "r", encoding="utf-8", errors="replace") as f:
return f.read()
return None
def cache_put(cache_dir: str, key: str, fasta: str) -> None:
if not cache_dir:
return
os.makedirs(cache_dir, exist_ok=True)
path = os.path.join(cache_dir, f"{key}.fasta")
with open(path, "w", encoding="utf-8") as f:
f.write(fasta)
def rewrite_fasta_header(fasta: str, new_header: str) -> str:
lines = fasta.splitlines()
if not lines or not lines[0].startswith(">"):
return fasta
lines[0] = ">" + new_header
return "\n".join(lines) + ("\n" if not fasta.endswith("\n") else "")
def fetch_best_fasta(session: requests.Session, pid: str, cache_dir: str = "") -> Tuple[Optional[str], str, Optional[str]]:
"""
Returns (fasta_text, status, resolved_upi)
status: uniprotkb | uniparc | uniparc_direct | not_found | *_cache
"""
if pid.startswith("UPI"):
cached = cache_get(cache_dir, pid)
if cached:
return cached, "uniparc_direct_cache", pid
fasta = fetch_uniparc_fasta(session, pid)
if fasta:
cache_put(cache_dir, pid, fasta)
return fasta, "uniparc_direct", pid
return None, "not_found", pid
cached = cache_get(cache_dir, pid)
if cached:
return cached, "uniprotkb_cache", None
fasta = fetch_uniprotkb_fasta(session, pid)
if fasta:
cache_put(cache_dir, pid, fasta)
return fasta, "uniprotkb", None
upi = resolve_upi_via_uniparc_search(session, pid)
if not upi:
return None, "not_found", None
cached2 = cache_get(cache_dir, upi)
if cached2:
return cached2, "uniparc_cache", upi
fasta2 = fetch_uniparc_fasta(session, upi)
if fasta2:
cache_put(cache_dir, upi, fasta2)
return fasta2, "uniparc", upi
return None, "not_found", upi
def main():
ap = argparse.ArgumentParser()
ap.add_argument("--excel", required=True, help="Input Excel file")
ap.add_argument("--outdir", default="./exoproteome_fastas", help="Output folder")
ap.add_argument("--cache", default="./uniprot_cache", help="Cache folder for downloaded FASTAs")
ap.add_argument("--sleep", type=float, default=0.1, help="Sleep seconds between requests")
ap.add_argument("--sheets", nargs="*", default=DEFAULT_SHEETS, help="Sheet names to process")
args = ap.parse_args()
os.makedirs(args.outdir, exist_ok=True)
os.makedirs(args.cache, exist_ok=True)
log_path = os.path.join(args.outdir, "run.log")
def log(msg: str):
print(msg)
with open(log_path, "a", encoding="utf-8") as f:
f.write(msg + "\n")
with open(log_path, "w", encoding="utf-8") as f:
f.write("")
xls = pd.ExcelFile(args.excel)
missing = [s for s in args.sheets if s not in xls.sheet_names]
if missing:
log("WARNING: Missing sheets in workbook:")
for s in missing:
log(f" - {s}")
log("Continuing with available sheets only.\n")
session = requests.Session()
session.headers.update({"User-Agent": "exoproteome-fasta-fetch/3.0"})
for sheet in args.sheets:
if sheet not in xls.sheet_names:
continue
sheet_tag = sanitize_name(sheet)
out_fasta = os.path.join(args.outdir, f"{sheet_tag}.fasta")
out_map = os.path.join(args.outdir, f"{sheet_tag}.mapping.tsv")
out_up = os.path.join(args.outdir, f"{sheet_tag}.up.ids.txt")
out_down = os.path.join(args.outdir, f"{sheet_tag}.down.ids.txt")
tbl = extract_ids_and_fc_from_sheet(args.excel, sheet)
log(f"\n=== Sheet: {sheet} ===")
log(f"Extracted IDs (unique): {len(tbl)}")
if tbl.empty:
log("No IDs found. Skipping.")
continue
tbl["direction"] = [classify_direction_no_cutoff(x) for x in tbl["log2fc"].tolist()]
n_fc = int(tbl["log2fc"].notna().sum())
n_up = int((tbl["direction"] == "Up").sum())
n_dn = int((tbl["direction"] == "Down").sum())
n_zero = int((tbl["direction"] == "Zero").sum())
n_na = int((tbl["direction"] == "NA").sum())
log(f"log2FC present: {n_fc}/{len(tbl)}")
log(f"Direction (no cutoff): Up={n_up}, Down={n_dn}, Zero={n_zero}, NA={n_na}")
mapping_rows = []
fasta_chunks = []
n_ok = 0
n_nf = 0
for i, row in enumerate(tbl.itertuples(index=False), start=1):
pid = row.protein_id
log2fc = row.log2fc
direction = row.direction
fasta, status, upi = fetch_best_fasta(session, pid, cache_dir=args.cache)
if fasta:
header = pid
if upi and upi != pid:
header = f"{pid}|UniParc:{upi}"
fasta = rewrite_fasta_header(fasta, header)
fasta_chunks.append(fasta)
n_ok += 1
else:
n_nf += 1
mapping_rows.append({
"sheet": sheet,
"protein_id_input": pid,
"resolved_upi": upi if upi else "",
"status": status,
"log2fc": "" if log2fc is None else log2fc,
"direction": direction,
"fasta_written": "yes" if fasta else "no",
})
if args.sleep > 0:
time.sleep(args.sleep)
if i % 50 == 0:
log(f" progress: {i}/{len(tbl)} (ok={n_ok}, not_found={n_nf})")
with open(out_fasta, "w", encoding="utf-8") as f:
for chunk in fasta_chunks:
f.write(chunk.strip() + "\n")
map_df = pd.DataFrame(mapping_rows)
map_df.to_csv(out_map, sep="\t", index=False)
up_ids = map_df.loc[map_df["direction"] == "Up", "protein_id_input"].astype(str).tolist()
dn_ids = map_df.loc[map_df["direction"] == "Down", "protein_id_input"].astype(str).tolist()
with open(out_up, "w", encoding="utf-8") as f:
f.write("\n".join(up_ids) + ("\n" if up_ids else ""))
with open(out_down, "w", encoding="utf-8") as f:
f.write("\n".join(dn_ids) + ("\n" if dn_ids else ""))
log(f"Fetched FASTA: {n_ok}/{len(tbl)} (not_found={n_nf})")
log(f"Saved FASTA: {out_fasta}")
log(f"Saved mapping: {out_map}")
log(f"Saved Up IDs: {out_up} ({len(up_ids)})")
log(f"Saved Down IDs:{out_down} ({len(dn_ids)})")
log("\nDONE.")
if __name__ == "__main__":
main()
02_normalize_fasta_headers.py
#!/usr/bin/env python3
import re
import sys
from collections import defaultdict
def canon_id(header: str) -> str:
h = header.strip()
h = h[1:] if h.startswith(">") else h
# tr|ACC|NAME or sp|ACC|NAME
m = re.match(r'^(sp|tr)\|([^|]+)\|', h)
if m:
return m.group(2)
# otherwise: first token, then before first '|'
first = h.split()[0]
return first.split("|")[0]
def read_fasta(path):
with open(path, "r", encoding="utf-8", errors="replace") as f:
header = None
seq = []
for line in f:
line = line.rstrip("\n")
if line.startswith(">"):
if header is not None:
yield header, "".join(seq)
header = line
seq = []
else:
seq.append(line.strip())
if header is not None:
yield header, "".join(seq)
def main():
if len(sys.argv) < 3:
print("Usage: normalize_fasta_headers.py in.fasta out.fasta [map.tsv]", file=sys.stderr)
sys.exit(1)
in_fa = sys.argv[1]
out_fa = sys.argv[2]
map_tsv = sys.argv[3] if len(sys.argv) >= 4 else out_fa + ".idmap.tsv"
seen = defaultdict(int)
with open(out_fa, "w", encoding="utf-8") as fo, open(map_tsv, "w", encoding="utf-8") as fm:
fm.write("old_header\tnew_id\n")
for old_h, seq in read_fasta(in_fa):
base = canon_id(old_h)
seen[base] += 1
new_id = base if seen[base] == 1 else f"{base}__dup{seen[base]}"
fm.write(f"{old_h}\t{new_id}\n")
fo.write(f">{new_id}\n")
# wrap sequence at 60
for i in range(0, len(seq), 60):
fo.write(seq[i:i+60] + "\n")
print(f"Wrote: {out_fa}")
print(f"Wrote: {map_tsv}")
if __name__ == "__main__":
main()
03_wrapper_3circles_venn_RNAseq_Exo_ProALFA.R
#!/usr/bin/env Rscript
# ============================================================
# 3-circle Venn: RNA-seq (sig) vs Exoproteome vs Proximity/ALFApulldown
#
# Key points:
# - RNA-seq inputs are Excel files with sheet 'Complete_Data'
# * GeneID is the ID used for comparison (locus tag style)
# * Keep Preferred_name / Description for annotation in ALL Venn regions
# - Exoproteome uses MS protein IDs + log2FC sign (no cutoff), mapped to locus tags via BLAST best-hit TSV
# - Proximity/ALFApulldown (ProALFA) uses SIP/MS protein IDs mapped to locus tags via BLAST best-hit TSV
# - Debug stats + parsing/mapping summaries are written into a LOG file
#
# NOTE: (记得!) 生成 Exoproteome_ALL.fasta + Exoproteome_ALL_vs_ref.blast.tsv:
# cat Exoproteome_MH_1h.fasta Exoproteome_MH_4h.fasta Exoproteome_MH_18h.fasta \\
# Exoproteome_TSB_1h.fasta Exoproteome_TSB_4h.fasta Exoproteome_TSB_18h.fasta \\
# > Exoproteome_ALL.fasta
# blastp -query Exoproteome_ALL.fasta -db CP020463_protein.dmnd \\
# -out Exoproteome_ALL_vs_ref.blast.tsv -outfmt 6 -evalue 1e-5 -max_target_seqs 50 -num_threads 40
# ============================================================
suppressPackageStartupMessages({
library(tidyverse)
library(readxl)
library(openxlsx)
library(VennDiagram)
library(grid)
})
# -------------------------
# CONFIG
# -------------------------
RNA_PADJ_CUT <- 0.05
RNA_LFC_CUT <- 2
OUTDIR <- "./venn_outputs"
dir.create(OUTDIR, showWarnings = FALSE, recursive = TRUE)
LOGFILE <- file.path(OUTDIR, "pipeline_log.txt")
sink(LOGFILE, split = TRUE)
cat("=== 3-circle Venn pipeline ===\n")
cat(sprintf("RNA cutoff: padj < %.3g and |log2FoldChange| > %.3g\n", RNA_PADJ_CUT, RNA_LFC_CUT))
cat("Output dir:", normalizePath(OUTDIR), "\n\n")
# RNA inputs (Excel, sheet = Complete_Data)
RNA_FILES <- list(
deltasbp_MH_2h = "./DEG_Annotations_deltasbp_MH_2h_vs_WT_MH_2h-all.xlsx",
deltasbp_MH_4h = "./DEG_Annotations_deltasbp_MH_4h_vs_WT_MH_4h-all.xlsx",
deltasbp_MH_18h = "./DEG_Annotations_deltasbp_MH_18h_vs_WT_MH_18h-all.xlsx",
deltasbp_TSB_2h = "./DEG_Annotations_deltasbp_TSB_2h_vs_WT_TSB_2h-all.xlsx",
deltasbp_TSB_4h = "./DEG_Annotations_deltasbp_TSB_4h_vs_WT_TSB_4h-all.xlsx",
deltasbp_TSB_18h = "./DEG_Annotations_deltasbp_TSB_18h_vs_WT_TSB_18h-all.xlsx"
)
# Exoproteome sheets (from summary Excel)
EXO_SUMMARY_XLSX <- "./Zusammenfassung SIP und Exoproteome.xlsx"
EXO_SHEETS <- list(
Exoproteome_MH_1h = "Exoproteome MH 1h",
Exoproteome_MH_4h = "Exoproteome MH 4h",
Exoproteome_MH_18h = "Exoproteome MH 18h",
Exoproteome_TSB_1h = "Exoproteome TSB 1h",
Exoproteome_TSB_4h = "Exoproteome TSB 4h",
Exoproteome_TSB_18h = "Exoproteome TSB 18h"
)
# Exoproteome BLAST best-hit mapping (query protein -> locus tag)
EXO_BLAST_TSV <- "./Exoproteome_ALL_vs_ref.besthit.tsv"
# Proximity/ALFApulldown sheets (from same summary Excel)
PROALFA_SUMMARY_XLSX <- "./Zusammenfassung SIP und Exoproteome.xlsx"
PROALFA_SHEETS <- list(
Proximity_4h = "Proximity 4h",
Proximity_18h = "Proximity 18h",
ALFApulldown_4h = "ALFApulldown 4h",
ALFApulldown_18h = "ALFApulldown 18h"
)
# ProALFA BLAST best-hit mapping (each dataset protein -> locus tag)
PROALFA_BLAST_TSV <- list(
Proximity_4h = "./Proximity_4h_vs_ref.besthit.tsv",
Proximity_18h = "./Proximity_18h_vs_ref.besthit.tsv",
ALFApulldown_4h = "./ALFApulldown_4h_vs_ref.besthit.tsv",
ALFApulldown_18h = "./ALFApulldown_18h_vs_ref.besthit.tsv"
)
# FASTA inputs (QC only)
FASTA_FILES <- list(
Proximity_4h = "./Proximity_4h.fasta",
Proximity_18h = "./Proximity_18h.fasta",
ALFApulldown_4h = "./ALFApulldown_4h.fasta",
ALFApulldown_18h = "./ALFApulldown_18h.fasta",
Exo_MH_1h = "./Exoproteome_MH_1h.fasta",
Exo_MH_4h = "./Exoproteome_MH_4h.fasta",
Exo_MH_18h = "./Exoproteome_MH_18h.fasta",
Exo_TSB_1h = "./Exoproteome_TSB_1h.fasta",
Exo_TSB_4h = "./Exoproteome_TSB_4h.fasta",
Exo_TSB_18h = "./Exoproteome_TSB_18h.fasta"
)
# -------------------------
# HELPERS
# -------------------------
safe_file_check <- function(fp, label) {
if (!file.exists(fp)) stop(label, " missing: ", fp)
}
canonical_id_one <- function(x) {
x <- as.character(x)
x <- stringr::str_trim(x)
if (is.na(x) || x == "") return(NA_character_)
# remove leading ">"
x <- stringr::str_remove(x, "^>")
# keep first token before whitespace
x <- stringr::str_split(x, "\\s+", simplify = TRUE)[1]
# handle UniProt-style: tr|ID|... or sp|ID|...
if (stringr::str_detect(x, "\\|")) {
parts <- stringr::str_split(x, "\\|")[[1]]
if (length(parts) >= 2 && parts[1] %in% c("tr", "sp")) {
x <- parts[2]
} else {
x <- parts[1]
}
}
x
}
canonical_id <- function(x) {
vapply(x, canonical_id_one, character(1))
}
read_fasta_headers <- function(fp) {
safe_file_check(fp, "FASTA")
hdr <- readLines(fp, warn = FALSE) %>% keep(~str_starts(.x, ">"))
tibble(raw = hdr, id = canonical_id(hdr)) %>%
filter(!is.na(id), id != "")
}
norm_name <- function(x) {
x <- tolower(trimws(as.character(x)))
x <- gsub("[^a-z0-9]+", "", x)
x
}
# Robust column picker (case-insensitive + punctuation-insensitive)
find_col <- function(nms, target) {
nms <- as.character(nms)
target_n <- norm_name(target)
n_n <- norm_name(nms)
# exact normalized match
idx <- which(n_n == target_n)
if (length(idx) > 0) return(nms[idx[1]])
# contains (normalized)
idx <- which(str_detect(n_n, fixed(target_n)))
if (length(idx) > 0) return(nms[idx[1]])
NA_character_
}
# Best-hit parser (BLAST outfmt6):
# qid sid pident length mismatch gapopen qstart qend sstart send evalue bitscore
read_besthit_tsv <- function(fp) {
safe_file_check(fp, "BLAST TSV")
suppressPackageStartupMessages({
df <- read_tsv(fp, col_names = FALSE, show_col_types = FALSE, progress = FALSE)
})
if (ncol(df) < 12) stop("BLAST TSV has <12 columns: ", fp)
colnames(df)[1:12] <- c("qid","sid","pident","length","mismatch","gapopen",
"qstart","qend","sstart","send","evalue","bitscore")
df %>%
mutate(
qid = canonical_id(qid),
sid = as.character(sid),
bitscore = suppressWarnings(as.numeric(bitscore)),
evalue = suppressWarnings(as.numeric(evalue))
) %>%
filter(!is.na(qid), qid != "")
}
besthit_reduce <- function(df) {
# one best hit per query: highest bitscore, then lowest evalue
df %>%
arrange(qid, desc(bitscore), evalue) %>%
group_by(qid) %>%
slice(1) %>%
ungroup()
}
# Try to extract locus tag from subject id
extract_locus_tag <- function(sid) {
sid <- as.character(sid)
# if sid already looks like B4U56_00090
if (str_detect(sid, "^B4U56_\\d{5}$")) return(sid)
# otherwise try to find locus tag pattern anywhere
hit <- str_extract(sid, "B4U56_\\d{5}")
hit
}
read_exo_sheet <- function(xlsx, sheet) {
df <- read_excel(xlsx, sheet = sheet, col_names = TRUE)
# Protein column: assume first column contains protein IDs
prot_col <- names(df)[1]
# log2FC column: per user it's column D, name looks like "log 2 FC)"
log2_col <- find_col(names(df), "log 2 FC)")
if (is.na(log2_col)) {
# fallback fuzzy: any col containing both log2 and fc
hits <- names(df)[str_detect(norm_name(names(df)), "log2") & str_detect(norm_name(names(df)), "fc")]
if (length(hits) > 0) log2_col <- hits[1]
}
if (is.na(log2_col)) stop("Exoproteome sheet missing log2FC column ('log 2 FC)'): ", sheet)
out <- df %>%
transmute(
Protein_raw = as.character(.data[[prot_col]]),
log2FC = suppressWarnings(as.numeric(.data[[log2_col]]))
) %>%
filter(!is.na(Protein_raw), Protein_raw != "")
out <- out %>%
mutate(
Protein_ID = canonical_id(Protein_raw),
EXO_direction = case_when(
is.na(log2FC) ~ NA_character_,
log2FC > 0 ~ "UP",
log2FC < 0 ~ "DOWN",
TRUE ~ "ZERO"
)
) %>%
filter(!is.na(Protein_ID), Protein_ID != "")
out
}
read_rna_excel <- function(fp) {
safe_file_check(fp, "RNA Excel")
df <- read_excel(fp, sheet = "Complete_Data", col_names = TRUE)
# required columns
req <- c("GeneID","padj","log2FoldChange")
miss <- setdiff(req, names(df))
if (length(miss) > 0) stop("RNA Excel missing columns: ", paste(miss, collapse=", "), " in ", fp)
# optional annotation columns
pref_col <- if ("Preferred_name" %in% names(df)) "Preferred_name" else NA_character_
desc_col <- if ("Description" %in% names(df)) "Description" else NA_character_
df <- df %>%
mutate(
GeneID_plain = str_remove(as.character(GeneID), "^gene-"),
padj = suppressWarnings(as.numeric(padj)),
log2FoldChange = suppressWarnings(as.numeric(log2FoldChange)),
Preferred_name = if (!is.na(pref_col)) as.character(.data[[pref_col]]) else NA_character_,
Description = if (!is.na(desc_col)) as.character(.data[[desc_col]]) else NA_character_
)
df
}
# -------------------------
# FASTA QC
# -------------------------
cat("[FASTA] Reading FASTA files for QC (10)\n")
fasta_qc_stats <- list()
for (nm in names(FASTA_FILES)) {
fp <- FASTA_FILES[[nm]]
safe_file_check(fp, "FASTA file")
h <- read_fasta_headers(fp)
n_hdr <- nrow(h)
n_uniq <- n_distinct(h$id)
n_dup <- n_hdr - n_uniq
cat(sprintf(" - %s: headers=%d, unique_IDs=%d, duplicates=%d\n", nm, n_hdr, n_uniq, n_dup))
dup_ids <- h %>% count(id, sort = TRUE) %>% filter(n > 1)
fasta_qc_stats[[nm]] <- tibble(
Dataset = nm,
headers = n_hdr,
unique_IDs = n_uniq,
duplicates = n_dup,
top_duplicate_id = ifelse(nrow(dup_ids) > 0, dup_ids$id[1], NA_character_),
top_duplicate_n = ifelse(nrow(dup_ids) > 0, dup_ids$n[1], NA_integer_)
)
}
fasta_qc_stats_df <- bind_rows(fasta_qc_stats)
# -------------------------
# RNA: read 6 comparisons (Excel)
# -------------------------
cat("\n[RNA] Reading 6 RNA-seq comparisons (Excel: Complete_Data)\n")
rna_sig_long <- list()
rna_all_long <- list()
for (cmp in names(RNA_FILES)) {
fp <- RNA_FILES[[cmp]]
df <- read_rna_excel(fp)
# Keep a full-gene annotation table (used later to annotate EXO/ProALFA-only members)
rna_all_long[[cmp]] <- df %>%
mutate(Comparison = cmp) %>%
select(GeneID_plain, Preferred_name, Description, padj, log2FoldChange, Comparison)
# Debug: how many bad entries
bad_gene <- sum(is.na(df$GeneID_plain) | df$GeneID_plain == "")
bad_padj <- sum(is.na(df$padj))
bad_lfc <- sum(is.na(df$log2FoldChange))
sig <- df %>%
filter(!is.na(GeneID_plain), GeneID_plain != "",
!is.na(padj), !is.na(log2FoldChange),
padj < RNA_PADJ_CUT,
abs(log2FoldChange) > RNA_LFC_CUT) %>%
mutate(
Comparison = cmp,
RNA_direction = ifelse(log2FoldChange > 0, "UP", "DOWN")
) %>%
select(GeneID_plain, Comparison, padj, log2FoldChange, RNA_direction,
Preferred_name, Description)
cat(sprintf(" - %s: rows=%d (bad GeneID=%d, bad padj=%d, bad log2FC=%d); significant=%d (UP=%d, DOWN=%d)\n",
cmp, nrow(df), bad_gene, bad_padj, bad_lfc,
nrow(sig), sum(sig$RNA_direction == "UP"), sum(sig$RNA_direction == "DOWN")))
rna_sig_long[[cmp]] <- sig
}
rna_sig_long <- bind_rows(rna_sig_long)
rna_all_long <- bind_rows(rna_all_long)
RNA <- sort(unique(rna_sig_long$GeneID_plain))
cat(sprintf("[RNA] Union significant genes (6 comps): %d\n", length(RNA)))
# Build an annotation lookup from *ALL* genes (so EXO-only/ProALFA-only also get annotation)
rna_ann <- rna_all_long %>%
filter(!is.na(GeneID_plain), GeneID_plain != "") %>%
group_by(GeneID_plain) %>%
summarise(
Preferred_name = coalesce(Preferred_name[match(TRUE, !is.na(Preferred_name) & Preferred_name != "", !is.na(Preferred_name) & Preferred_name != "")], Preferred_name[1]),
Description = coalesce(Description[match(TRUE, !is.na(Description) & Description != "", !is.na(Description) & Description != "")], Description[1]),
.groups = "drop"
)
# -------------------------
# EXO: read exoproteome sheets + mapping (BLAST)
# -------------------------
cat("\n[EXO] Reading Exoproteome sheets + mapping via BLAST\n")
safe_file_check(EXO_SUMMARY_XLSX, "Exoproteome summary Excel")
safe_file_check(EXO_BLAST_TSV, "Exoproteome BLAST TSV")
exo_long <- list()
for (sh in names(EXO_SHEETS)) {
df <- read_exo_sheet(EXO_SUMMARY_XLSX, EXO_SHEETS[[sh]]) %>%
mutate(Sheet = EXO_SHEETS[[sh]])
cat(sprintf(" - %s: proteins=%d (with log2FC=%d; UP=%d, DOWN=%d)\n",
EXO_SHEETS[[sh]],
n_distinct(df$Protein_ID),
sum(!is.na(df$log2FC)),
sum(df$EXO_direction == "UP", na.rm = TRUE),
sum(df$EXO_direction == "DOWN", na.rm = TRUE)))
exo_long[[sh]] <- df
}
exo_long <- bind_rows(exo_long) %>%
distinct(Protein_ID, Sheet, .keep_all = TRUE)
# BLAST mapping
exo_blast <- read_besthit_tsv(EXO_BLAST_TSV)
exo_best <- besthit_reduce(exo_blast) %>%
mutate(LocusTag = map_chr(sid, extract_locus_tag))
cat(sprintf("[BLAST] Exoproteome_ALL rows=%d, queries=%d, besthits=%d, unique_targets=%d\n",
nrow(exo_blast), n_distinct(exo_blast$qid), nrow(exo_best), n_distinct(exo_best$LocusTag)))
dup_targets <- exo_best %>%
filter(!is.na(LocusTag)) %>%
count(LocusTag, sort = TRUE) %>%
filter(n > 1)
cat(sprintf("[BLAST] Exoproteome_ALL duplicated targets in besthits: %d (top 5)\n", nrow(dup_targets)))
if (nrow(dup_targets) > 0) print(head(dup_targets, 5))
exo_mapped <- exo_long %>%
left_join(exo_best %>% select(qid, LocusTag), by = c("Protein_ID" = "qid")) %>%
mutate(LocusTag = as.character(LocusTag))
cat(sprintf("[EXO] Proteins before mapping: %d unique\n", n_distinct(exo_long$Protein_ID)))
cat(sprintf("[EXO] Mapped proteins (have LocusTag): %d\n", sum(!is.na(exo_mapped$LocusTag))))
cat(sprintf("[EXO] Unique mapped proteins: %d\n", n_distinct(exo_mapped$Protein_ID[!is.na(exo_mapped$LocusTag)])))
cat(sprintf("[EXO] Unique mapped locus tags: %d\n", n_distinct(exo_mapped$LocusTag[!is.na(exo_mapped$LocusTag)])))
EXO <- sort(unique(na.omit(exo_mapped$LocusTag)))
# -------------------------
# ProALFA: read proximity + ALFA sheets + mapping (BLAST)
# -------------------------
cat("\n[ProALFA] Reading Proximity/ALFA sheets + mapping via BLAST\n")
safe_file_check(PROALFA_SUMMARY_XLSX, "ProALFA summary Excel")
ms_long <- list()
for (ds in names(PROALFA_SHEETS)) {
sh <- PROALFA_SHEETS[[ds]]
if (!(sh %in% readxl::excel_sheets(PROALFA_SUMMARY_XLSX))) {
stop("Sheet '", sh, "' not found")
}
df <- read_excel(PROALFA_SUMMARY_XLSX, sheet = sh, col_names = TRUE)
# assume first column has protein IDs
prot_col <- names(df)[1]
tmp <- df %>%
transmute(
Protein_raw = as.character(.data[[prot_col]]),
Protein_ID = canonical_id(Protein_raw)
) %>%
filter(!is.na(Protein_ID), Protein_ID != "") %>%
distinct(Protein_ID)
cat(sprintf(" - %s (%s): proteins=%d unique\n", ds, sh, nrow(tmp)))
ms_long[[ds]] <- tmp %>% mutate(Dataset = ds)
}
ms_long <- bind_rows(ms_long)
# mapping per dataset
ms_mapped_list <- list()
PROALFA_sets <- list()
for (ds in names(PROALFA_BLAST_TSV)) {
bt <- PROALFA_BLAST_TSV[[ds]]
safe_file_check(bt, paste0("ProALFA BLAST TSV: ", ds))
b <- read_besthit_tsv(bt)
best <- besthit_reduce(b) %>%
mutate(LocusTag = map_chr(sid, extract_locus_tag))
cat(sprintf("[BLAST] %s rows=%d, queries=%d, besthits=%d, unique_targets=%d\n",
ds, nrow(b), n_distinct(b$qid), nrow(best), n_distinct(best$LocusTag)))
dup_t <- best %>%
filter(!is.na(LocusTag)) %>%
count(LocusTag, sort = TRUE) %>%
filter(n > 1)
if (nrow(dup_t) > 0) {
cat(sprintf("[BLAST] %s duplicated targets in besthits: %d (top 5)\n", ds, nrow(dup_t)))
print(head(dup_t, 5))
}
tmp_ds <- ms_long %>%
filter(Dataset == ds) %>%
left_join(best %>% select(qid, LocusTag), by = c("Protein_ID" = "qid"))
cat(sprintf("[ProALFA] %s: proteins=%d, mapped=%d, unique_targets=%d\n",
ds,
n_distinct(tmp_ds$Protein_ID),
sum(!is.na(tmp_ds$LocusTag)),
n_distinct(tmp_ds$LocusTag[!is.na(tmp_ds$LocusTag)])))
ms_mapped_list[[ds]] <- tmp_ds
PROALFA_sets[[ds]] <- sort(unique(na.omit(tmp_ds$LocusTag)))
}
ms_mapped_all <- bind_rows(ms_mapped_list)
ProALFA <- sort(unique(unlist(PROALFA_sets)))
cat(sprintf("[ProALFA] Union locus tags (Proximity+ALFA): %d\n", length(ProALFA)))
# -------------------------
# VENN intersections
# -------------------------
cat("\n[VENN] Computing intersections\n")
cat(sprintf("Set sizes: RNA=%d, EXO=%d, ProALFA=%d\n", length(RNA), length(EXO), length(ProALFA)))
i_RNA_EXO <- intersect(RNA, EXO)
i_RNA_ProALFA <- intersect(RNA, ProALFA)
i_EXO_ProALFA <- intersect(EXO, ProALFA)
i_all3 <- Reduce(intersect, list(RNA, EXO, ProALFA))
cat(sprintf("Intersections: RNA∩EXO=%d, RNA∩ProALFA=%d, EXO∩ProALFA=%d, ALL3=%d\n",
length(i_RNA_EXO), length(i_RNA_ProALFA), length(i_EXO_ProALFA), length(i_all3)))
# -------------------------
# Venn plot
# -------------------------
venn_png <- file.path(OUTDIR, "Venn_RNA_EXO_ProALFA.png")
venn_pdf <- file.path(OUTDIR, "Venn_RNA_EXO_ProALFA.pdf")
cat("[VENN] Writing plot:", venn_png, "\n")
venn.diagram(
x = list(RNA=RNA, Exoproteome=EXO, ProALFA=ProALFA),
category.names = c("RNA", "Exoproteome", "Proximity/ALFApulldown"),
filename = venn_png,
imagetype = "png",
height = 2400, width = 2400, resolution = 300,
fill = c("#4C72B0","#55A868","#C44E52"),
alpha = 0.4,
cex = 0.9,
cat.cex = 0.85,
cat.pos = c(-20, 20, 270),
# Increase 3rd value to push the long bottom label further out
cat.dist = c(0.06, 0.06, 0.16),
margin = 0.22,
main = "RNA-seq (sig) vs Exoproteome vs Proximity/ALFApulldown"
)
cat("[VENN] Writing plot:", venn_pdf, "\n")
vd <- venn.diagram(
x = list(RNA=RNA, Exoproteome=EXO, ProALFA=ProALFA),
category.names = c("RNA", "Exoproteome", "Proximity/ALFApulldown"),
filename = NULL,
fill = c("#4C72B0","#55A868","#C44E52"),
alpha = 0.4,
cex = 0.9,
cat.cex = 0.85,
cat.pos = c(-20, 20, 270),
cat.dist = c(0.06, 0.06, 0.16),
margin = 0.22,
main = "RNA-seq (sig) vs Exoproteome vs Proximity/ALFApulldown"
)
pdf(venn_pdf, width=7.5, height=7.5)
grid.draw(vd)
dev.off()
# -------------------------
# Region table (ALL members + direction annotations)
# -------------------------
all_members <- tibble(LocusTag = sort(unique(c(RNA, EXO, ProALFA)))) %>%
mutate(
in_RNA = LocusTag %in% RNA,
in_EXO = LocusTag %in% EXO,
in_ProALFA = LocusTag %in% ProALFA
)
# RNA direction per locus tag (from significant set only; union across comparisons)
rna_dir_tbl <- rna_sig_long %>%
group_by(GeneID_plain) %>%
summarise(
RNA_comparisons = paste(sort(unique(Comparison)), collapse = ";"),
RNA_dirs = paste(sort(unique(RNA_direction)), collapse = ";"),
min_padj = suppressWarnings(min(padj, na.rm = TRUE)),
max_abs_log2FC = suppressWarnings(max(abs(log2FoldChange), na.rm = TRUE)),
.groups = "drop"
) %>%
rename(LocusTag = GeneID_plain)
# EXO direction aggregated per locus tag
exo_dir_tbl <- exo_mapped %>%
filter(!is.na(LocusTag)) %>%
group_by(LocusTag) %>%
summarise(
EXO_sheets = paste(sort(unique(Sheet)), collapse = ";"),
EXO_dirs = paste(sort(unique(EXO_direction)), collapse = ";"),
EXO_proteinIDs = paste(sort(unique(Protein_ID)), collapse = ";"),
.groups = "drop"
)
# ProALFA membership aggregated per locus tag
proalfa_tbl <- ms_mapped_all %>%
filter(!is.na(LocusTag)) %>%
group_by(LocusTag) %>%
summarise(
ProALFA_datasets = paste(sort(unique(Dataset)), collapse = ";"),
ProALFA_proteinIDs = paste(sort(unique(Protein_ID)), collapse = ";"),
.groups = "drop"
)
region_tbl <- all_members %>%
left_join(rna_dir_tbl, by = "LocusTag") %>%
left_join(exo_dir_tbl, by = "LocusTag") %>%
left_join(proalfa_tbl, by = "LocusTag") %>%
left_join(rna_ann, by = c("LocusTag" = "GeneID_plain")) %>%
arrange(desc(in_RNA), desc(in_EXO), desc(in_ProALFA), LocusTag)
region_only <- function(inRNA, inEXO, inProALFA) {
region_tbl %>%
filter(in_RNA == inRNA, in_EXO == inEXO, in_ProALFA == inProALFA)
}
# -------------------------
# Excel output
# -------------------------
out_xlsx <- file.path(OUTDIR, "Venn_RNA_EXO_ProALFA_detailed.xlsx")
cat("[EXCEL] Writing:", out_xlsx, "\n")
wb <- createWorkbook()
addWorksheet(wb, "Summary")
writeData(wb, "Summary", data.frame(
Metric = c("RNA cutoff", "RNA union", "EXO union", "ProALFA union",
"RNA∩EXO", "RNA∩ProALFA", "EXO∩ProALFA", "ALL3"),
Value = c(
sprintf("padj < %.3g & |log2FC| > %.3g", RNA_PADJ_CUT, RNA_LFC_CUT),
length(RNA), length(EXO), length(ProALFA),
length(i_RNA_EXO), length(i_RNA_ProALFA), length(i_EXO_ProALFA), length(i_all3)
)
))
addWorksheet(wb, "FASTA_QC")
writeData(wb, "FASTA_QC", fasta_qc_stats_df)
addWorksheet(wb, "RNA_sig_long")
writeData(wb, "RNA_sig_long", rna_sig_long)
addWorksheet(wb, "RNA_all_annotations")
writeData(wb, "RNA_all_annotations", rna_all_long)
addWorksheet(wb, "EXO_proteins")
writeData(wb, "EXO_proteins", exo_long)
addWorksheet(wb, "EXO_mapped")
writeData(wb, "EXO_mapped", exo_mapped)
addWorksheet(wb, "ProALFA_proteins")
writeData(wb, "ProALFA_proteins", ms_long)
addWorksheet(wb, "ProALFA_mapped")
writeData(wb, "ProALFA_mapped", ms_mapped_all)
addWorksheet(wb, "All_members_with_dirs")
writeData(wb, "All_members_with_dirs", region_tbl)
addWorksheet(wb, "Only_RNA")
writeData(wb, "Only_RNA", region_only(TRUE, FALSE, FALSE))
addWorksheet(wb, "Only_EXO")
writeData(wb, "Only_EXO", region_only(FALSE, TRUE, FALSE))
addWorksheet(wb, "Only_ProALFA")
writeData(wb, "Only_ProALFA", region_only(FALSE, FALSE, TRUE))
addWorksheet(wb, "RNA_EXO_only")
writeData(wb, "RNA_EXO_only", region_only(TRUE, TRUE, FALSE))
addWorksheet(wb, "RNA_ProALFA_only")
writeData(wb, "RNA_ProALFA_only", region_only(TRUE, FALSE, TRUE))
addWorksheet(wb, "EXO_ProALFA_only")
writeData(wb, "EXO_ProALFA_only", region_only(FALSE, TRUE, TRUE))
addWorksheet(wb, "ALL3")
writeData(wb, "ALL3", region_only(TRUE, TRUE, TRUE))
saveWorkbook(wb, out_xlsx, overwrite = TRUE)
cat("\nDONE.\nOutputs:\n")
cat(" -", venn_png, "\n")
cat(" -", venn_pdf, "\n")
cat(" -", out_xlsx, "\n")
cat(" - LOG:", normalizePath(LOGFILE), "\n")
# dump warnings (so they go into log too)
if (!is.null(warnings())) {
cat("\n[WARNINGS]\n")
print(warnings())
}
sink()
Final Notes
- All comparisons are locus-tag consistent, enabling clean cross-omics integration.
- Exoproteome regulation is treated directionally only, by design.
- Logs and Excel outputs are intentionally verbose to support downstream inspection and reuse.
This pipeline is designed to be transparent, auditable, and extensible—ideal for iterative multi-omics analysis and figure generation.